
基于物化性质对嗜热蛋白的预测
2017-04-10 06:13:27刀福英陈欣欣
生物信息学 2017年1期
刀福英,陈欣欣,林 昊
(神经信息教育部重点实验室信息生物学中心(电子科技大学生命科学与技术学院),成都610054)
基于物化性质对嗜热蛋白的预测
刀福英,陈欣欣,林 昊*
(神经信息教育部重点实验室信息生物学中心(电子科技大学生命科学与技术学院),成都610054)
嗜热蛋白在高温下能保持稳定性和活性,是研究蛋白质热稳定性的理想模型,开发一个蛋白质热稳定性识别的方法将对蛋白质工程和蛋白质的设计很有帮助。目前的研究中,氨基酸的组成及其物化性质一直被认为和蛋白质的热稳定性相关。本研究筛选出可靠的数据集,包括915个嗜热蛋白和793个非嗜热蛋白。利用蛋白质氨基酸的物化性质和氨基酸的组成表征嗜热蛋白,将二肽氨基酸组成整合到9组氨基酸物化性质中使蛋白序列公式化。支持向量机5折叠交叉验证表明:当gap=0时,290个特征产生的精度最高,为92.74%。因此说明对于分析蛋白质的热稳定性,所建立的预测模型将是一个很有效的工具。
嗜热蛋白;热稳定性;伪氨基酸组分;氨基酸物化性质
嗜热和嗜冷微生物是两种重要的极端微生物, 存在于其中的嗜热和嗜冷酶是基础研究和工业应用的热点,它有助于认知蛋白质折叠、蛋白质结构和功能的关系以及设计用于极端环境的生物催化剂。随着第一个极端嗜热微生物Methanococcusjannaschii基因组的公布, 研究者通过比较基因组(蛋白质组)的方法对其稳定性机制进行了深入的探讨。近年来, 不少嗜冷微生物的基因组测序工作陆续完成, 使得对嗜热和嗜冷蛋白稳定性机理的研究不断深入。尽管研究者对上述极端蛋白稳定性机理的探讨较多,但利用蛋白质序列信息对其嗜热和嗜冷特性的理论预测却很少。
从蛋白质序列出发对其高级结构及特性进行理论预测所面临的一个重要课题是如何有效提取蛋白质序列特征, 氨基酸组成是最常用的一种方法, 此外, 利用二肽组成和伪氨基酸组成在一些情况下也取得了较好效果。在后基因组时代,随着DNA和蛋白质序列及结构信息的大量积累,人们利用数学、计算机科学的知识分析、挖掘生物数据,以寻求蕴涵在其中的生物学规律。
基于蛋白质序列特性可以对嗜热蛋白进行预测,Liang等[1]使用氨基酸耦合模型去区分嗜热与嗜常温蛋白,Zhang等[2]利用二肽和氨基酸组分来区分嗜热与嗜常温蛋白,其中五折交叉验证精度达86.6%,后来Gromiha和Suresh[3]将他们的数据去除冗余后,在神经网络的基础上运用氨基酸组分得到的五折交叉验证精度达到了89%。Montanucci等[4]运用支持向量机去预测蛋白质热稳定性,jackknife交叉检验的预测精度为88%。Wu等[5]提议运用决策树来预测蛋白质热稳定性,其预测精度在80%以上。尽管以上这些研究都获得了好的结果,但预测精度还有待提高。
在本文的研究中,构建了包括915个嗜热蛋白和793个非嗜热蛋白在内的很可靠的标准数据集,运用氨基酸二肽组分和九组氨基酸物化性质来表征蛋白质的特征,通过方差分析来进行特征筛选,利用支持向量机区分嗜热与非嗜热蛋白。本文使用的特征筛选技术可以提高预测精度,经过优化的290个参数的五折叠交叉验证准确率达到了92.74%,Jackknife交叉验证结果显示有91.69%的嗜热蛋白和91.42%的非嗜热蛋白是正确预测的,其ROC曲线面积为0.963。因此表明本文构建了较为精准的模型,可以通过对未知蛋白的序列预测其耐热性,从而可以判断其是否具有热稳定性,是否可以运用于相应的酶工程之中。
1 材料与方法
1.1 构建数据集
在当前的研究中,嗜热蛋白和非嗜热蛋白分别从嗜热有机体和非嗜热有机体中提取的。为了保证当温度上升到嗜热生物的温度时使获得的非嗜热蛋白变性,将60 ℃作为嗜热有机体最适生长温度的最低温度限制,将30 ℃作为非嗜热有机体最适生长温度的最高温度限制,对NCBI里1 126个全微生物基因组生物的最适生长温度进行筛选,有136个原核基因组(包括17个古生菌和119个细菌)满足要求。
从UniProt中根据最适温度的标记分别从136个原核有机体中选取嗜热和非嗜热蛋白序列,为了保证得到数据的可靠性则需要满足以下筛选步骤:(1)蛋白质必须是经过手动注释和审核的;(2)排除蛋白质序列中具有歧义的残基(例如带有“X”,“B”和“Z”);(3)排除含有其他蛋白片段的序列;(4)排除从预测或同源关系中推论的缺少可信度的蛋白质。严格遵照以上4个程序筛选得到1 329个嗜热蛋白和1 250个非嗜热蛋白。
这里构建的初步数据集中,通常还会存在一些冗余序列。数据集如果由许多相似度较高的样本组成,那么较高的冗余度就会导致统计代表性降低。如果预测器由一个有偏倚的数据集训练而来,更有可能产生错误的高估结果。为了除去冗余并避免偏倚,使用了CD-HIT软件[6]来筛选这些序列片段。
CD-HIT的基本思路是先对所有数据集里的序列,根据序列的长度从长到短进行排序,以最长的一条序列作为第一个序列类。然后依次处理排好序的各条序列,CD-HIT不仅能够对单独的数据集执行去除冗余信息,还可以比较两个不同的数据集。
本文选取的一致性阈值为0.4,去除序列相似度性在40%以上的序列后,最终的数据集包括915个嗜热蛋白和793个非嗜热蛋白,最终获得了1 708个样本作为基准数据集S,用公式表示如下:
S=ST∪Snon-T
(1)
这里的两个子集分别包含915个嗜热蛋白样本和793个非嗜热蛋白样本,符号“∪”表示两个子集的并集。
1.2 特征提取
在嗜热蛋白的预测中,用有效的数学公式来规定蛋白质序列是一个很有效的方法。一个很直接的办法是将公式表示全部蛋白质序列的全部氨基酸,公式如下:
P=R1R2R3…RL
(2)
其中R1、R2、R3......RL分别表示蛋白质样本P中的第1个,第2个,第3个,…… ,第L个氨基酸残基,有了这样的公式,就可以被很多序列相似搜索工具用来进行数据的预测,比如BLAST、FASTA等,对于一个高的相似序列的数据集,它的预测结果往往是很好的,所以这样的基于相似的方法是很直观的,但是有一个不可忽视的问题,在训练的数据集中查询序列的相似序列如果不能被找到的话它是就不会起作用。因此在对蛋白分类时提议利用离散向量表示蛋白质样本。伪氨基酸组分表示蛋白质序列是一个被广泛使用的方法,伪氨基酸组分(PseACC)[7]是一种能够很好地表征蛋白质序列的信息参数。它不但能够描述蛋白质序列的氨基酸组成,而且能够描述蛋白质氨基酸序列的物理化学性质的关联。基于伪氨基酸组分的概念,本文做了一个提升,将二肽氨基酸组分代替氨基酸组分,并且进行十组这样的特征提取,即gap值从0取到9,表示两氨基酸残基间间隔从0到9。
将g-gap二肽氨基酸组成来代替氨基酸组成,所以此参数不仅反映了两类蛋白在序列的组成和序列顺序的区别外,还能表现出残基间相关性,在基准数据集中将 400+nλ维向量表示每个蛋白质,表示公式如下:
P=[x1…x400x400+1…x400+nλ]T
(3)
其中
(4)
(5)
在公式(4)中,fu表示蛋白质P中二肽氨基酸的标准频率,公式(5)中nu表示蛋白质P中第u个二肽氨基酸的数量,很容易理解的二肽的数量总共有400(20×20)个,用二肽氨基酸频率来表征蛋白质的特征。
下面对氨基酸物化性质进行描述。公式(4)τu中的表示序列物化性质的相关性,由以下公式计算得到:
(6)
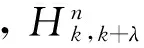
(7)
(8)
1.3 特征筛选
由公式(3)可知,用400+nλ个特征表示每个蛋白,为了能够得到最优的特征集,使用方差分析来进行特征筛选,将特征值进行排序,由以下公式来对特征打分:
(9)
在该公式中xu(i,j)表示在第i类样本中第j个样本的第u个特征的频率值;mi表示第i类样本的样本数(本文共有两类样本,m1=915为嗜热蛋白,m2=793为非嗜热蛋白)。分析该公式可知第u个特征对应的打分值F越大表明该特征区分嗜热蛋白与非嗜热蛋白的能力越强,因此将F值作为特征筛选标准。
1.4 支持向量机
根据耐热性对蛋白质进行预测就是蛋白质分类的过程。分类的方法很多,如费歇尔判别式,神经网络,集成学习,k-邻近算法等被广泛用于蛋白质的分类中。对小样本的分类本文使用支持向量机来构建分类器。
支持向量机(Support Vector Machine,SVM)[8]是目前极其流行的数据挖掘的工具。SVM的基本思想有如下两个方面:首先,支持向量机对线性条件下可以进行分类情况进行分析研究。当遇到线性条件下无法进行分类时,理论上应该把训练样本通过某种非线性的映射对数据进行升维处理,这样就会把数据升为较高维度的特征向量空间,在此空间中,寻找出线性的最佳超平面;其次,支持向量机的思想是建立在结构风险最小化的理论之上,支持向量机需要在高维空间中寻找分类超平面,寻找两种类别的样本点之间的最大分类间隔。本文通过网格搜索进行5折叠交叉验证,找到最佳的特征。支持向量机可以由libsvm软件包来运行。
1.5 评估指标
在统计学预测检验中,对于一个给定的基准数据集,jackknife检验[9]能够产生独一无二的结果,所以在实际应用中它经常被用来评估方法的性能。为了节省计算时间,本文在特征筛选的过程中使用5折叠交叉检验,挑选出最佳的特征集之后运用jackknife检验再次对特征集计算检验。它可在敏感性(Sn),特异性(Sp),准确率(Acc),马修相关系数(MCC)4个方面来评估。这4个参数由以下公式计算得到:
(10)
(11)
(12)
MCC=
(13)
Sn,Sp,Acc的范围为[0,1],MCC范围为[-1,1]。这里FN(False Negative)表示被判定为负样本,但事实上是正样本;FP(False Positive)表示被判定为正样本,但事实上是负样本;TN(True Negative)表示被判定为负样本,事实上也是负样本;TP(True Positive)表示被判定为正样本,事实上也是正样本。(本文正样本为嗜热蛋白,负样本为非嗜热蛋白)。这4个指标通常被用在统计预测理论中,它们可以从4个不同的角度来定量的衡量预测系统的性能。
此外,受试者特征曲线(ROC曲线)能兼顾灵敏度和特异性要求以综合评价分类器的预测性能,曲线下面积作为量化指标可以直观有效的比较不同分类器的性能优劣。线上的每个点都是对同一个分类器预测的反应,通常由于不同的判断标准得出了一系列不同的预测结果。受试者操作特征曲线的横坐标轴通常为虚报概率,纵坐标轴一般为击中概率,根据测试数据在特定分类器的不同的判断标准下得到的不同结果绘制出曲线。
2 结果与讨论
2.1 物化性质
在蛋白质的结构和功能中氨基酸的物化性质扮演着十分重要的角色,氨基酸的六种物化性质被广泛使用,分别是氨基酸的疏水性、亲水性、氨基酸侧链基团质量、-COOH基团的解离常数、-NH3基团解离常数、25℃时的等电点,在本文的研究中,除了以上六种物化性质外,还添加三种氨基酸物化性质,分别是氨基酸的刚性、柔性、不可替代性。九组氨基酸的物化性质[10]运用于公式(4)~(8)中。
在蛋白质的结构和功能中,氨基酸侧链基团的硬度和灵活性包含着重要有用的信息,刚性与柔性值是通过主成分分析获得的[11]。在生物的进化中,有些残基是很容易被替代的,但有些残基却很难被替代,不可替代性可由氨基酸的平均突变危险性来描述[12],平均突变危险性值越高表示该残基越难以被替代,不可替代性反应了在生命进化过程中的突变危险性。
2.2 预测精度
基于上面介绍的九组物化性质,本文可以得到400+9λ个特征,即在公式(3)~(6)中n=9,为了能够包含尽可能多的相关信息,节省计算资源,本文取λ=10,因此,用490维向量表示每个数据集中的每个蛋白质样本。
为得到最好的预测性能,挑选出具有最大精度的最佳特征,如果研究所有的特征,就会得到一个最好的特征集,但是490个特征的所有可能的组合的数目太大了,超出了大部分计算机的计算能力,所以要做到检验所有特征组合的性能那是不可能的,为节省计算时间,运用公式(9)中F打分来进行特征筛选,首先根据每个特征对应的F值从大到小进行排序,然后将第一个特征即具有最大F值的特征用SVM计算其精度,接下来,按照F值从大到小对应的特征值依次加到前一个特征集,依次每次进行SVM计算该特征集的精度,这个过程要一直重复,直到最小F值特征包含到该特征集中,即一共包含490个特征。所以最后SVM计算会产生相应的490个精度,分别是按照F值从大到小排列后的第一个特征对应的精度,前两个特征对应的精度,前三个特征对应的精度直到得到490个特征对应的精度为止,比较得到的精度,会得到一个最高精度对应的特征集。基于特征筛选技术,高维数据将会投射到低维空间,本文用该最佳的特征集来构建最终的预测模型。
变化参数gap的值分别取0到9,所以需要计算4 900(490×10)个特征集对应的精度,将特征数作为横坐标,将精度作为纵坐标,在笛卡尔坐标系中得到10组曲线图。如图1所示,当gap=0,横坐标290特征对应的精度为92.74%,该精度为最高精度。用jackknife检验计算该包含290个特征的模型,得Sn=91.69%,Sp=91.42%,表明该模型能够正确识别嗜热蛋白。
为了用这290个特征一目了然的描绘该模型的性能,在图2中绘制了ROC曲线,从图中可以看出曲线靠近左边和顶部坐标轴,表明该模型适用于嗜热蛋白与非嗜热蛋白的分类,在jackknife交叉检验中ROC曲线下的面积值为0.963。
为了对比,基于相同的数据集,还通过WEKA用了朴素贝叶斯[13]、贝叶斯网络、随机森林[14-15]三种方法进一步计算分类性能,预测结果显示在表1中,比较表1中的数据,很明显可以看出SVM是预测嗜热蛋白的最好的算法。
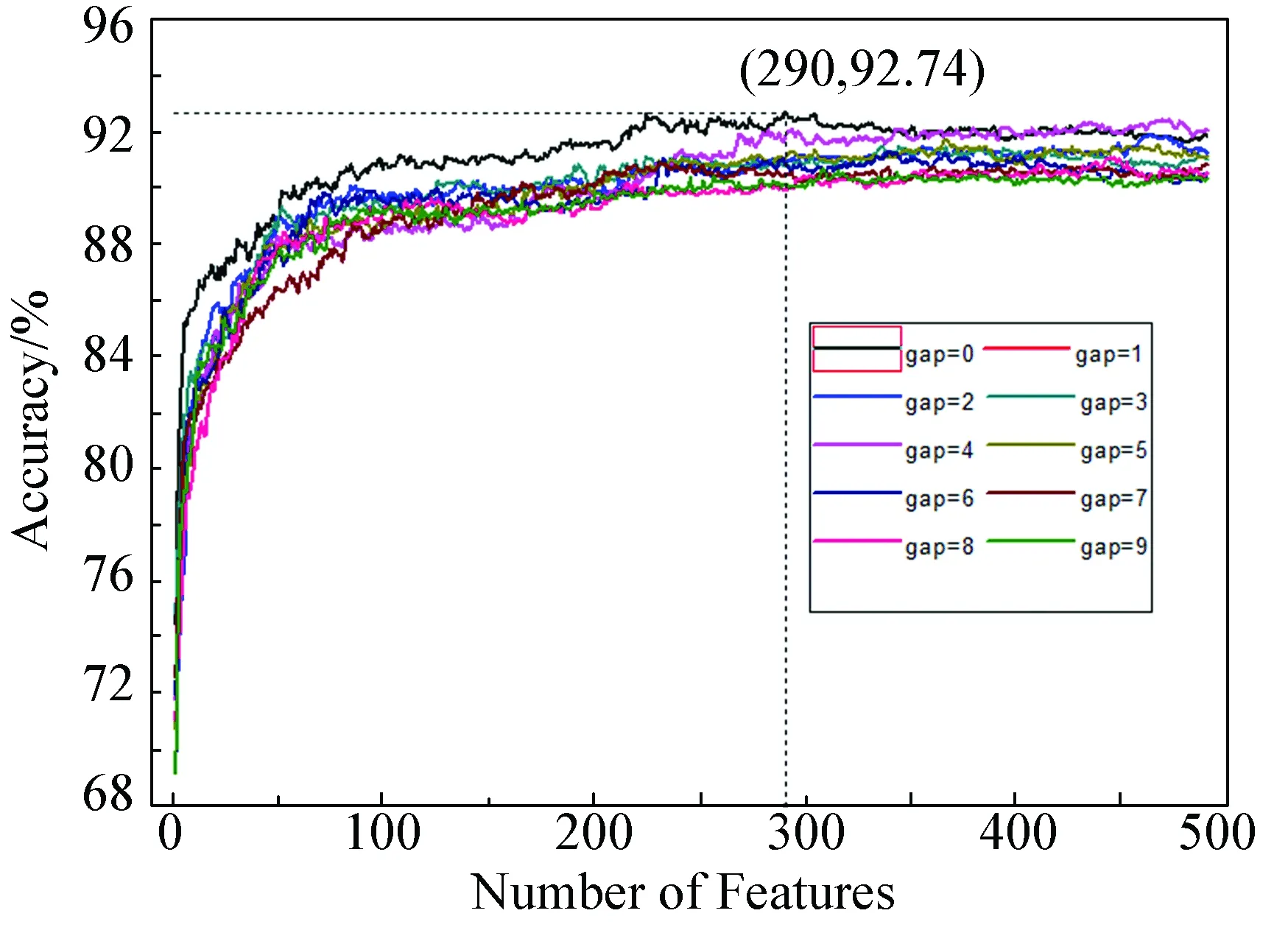
图1 特征筛选结果*Fig.1 A plot to show the feature selection results
*彩图见电子版(http://swxxx.alljournals.cn/ch/index.aspx)(2017年第1期DOI:10.3969/j.issn.1672-5565.2017.01.201606001)
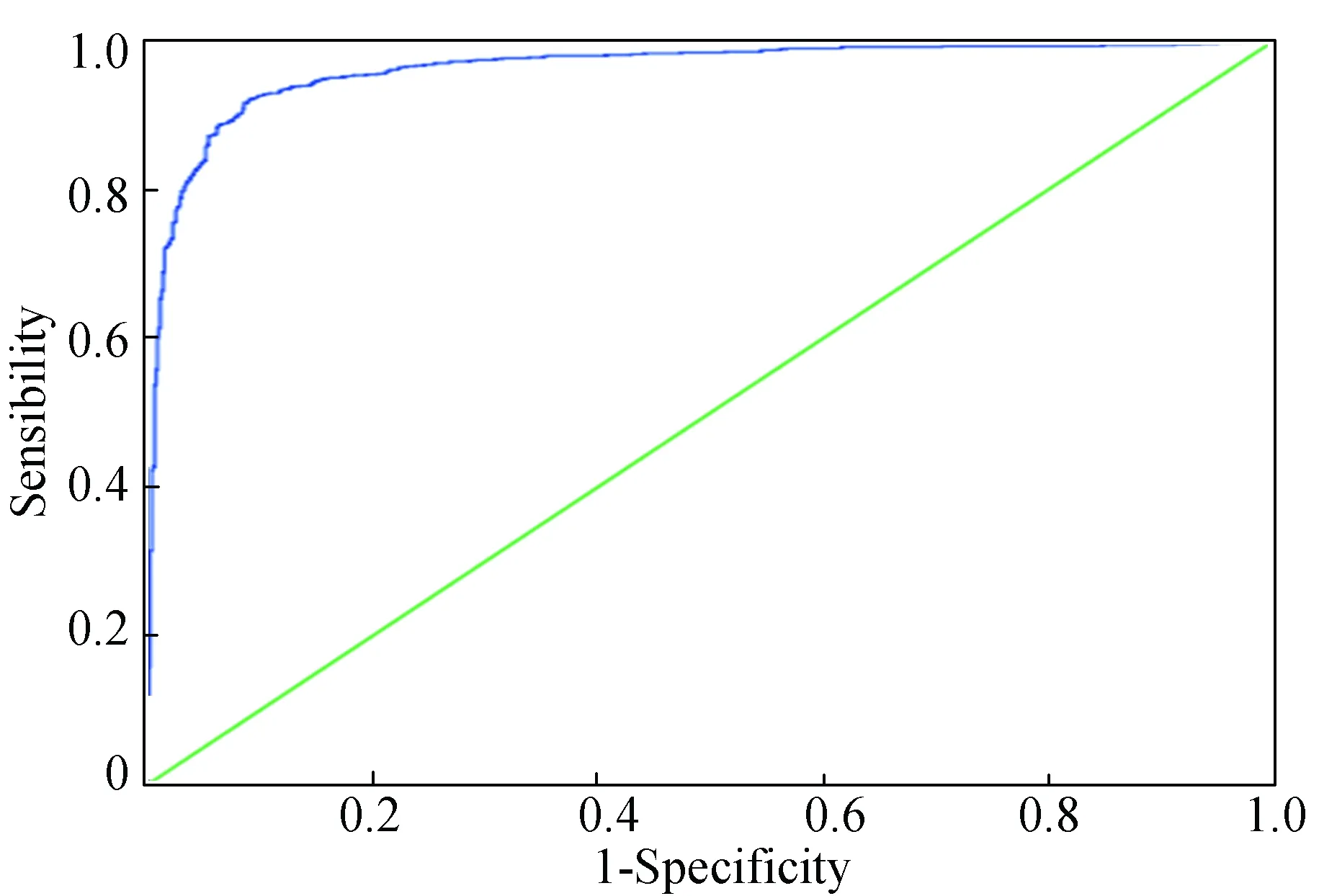
图2 最佳的290个特征在jackknife交叉验证中的ROC曲线Fig.2 The ROC curve for the model with 290 optimal 0-gap dipeptides in the jackknife cross-validation
注:对角线表示ROC的面积为0.5。

表1 比较不同算法的结果Table 1 Comparing the performance of different algorithms
3 总结与展望
蛋白质的热稳定性与酶工程密切相关,对蛋白质热稳定性的研究将对蛋白质工程和蛋白质的设计很有帮助,因此,开发了一个从非嗜热蛋白中筛选识别出嗜热蛋白的方法,获得了高精度的模型。结果表明,该方法可以筛选有效的特征,提高预测性能,在优化模型的基础上,将建立一个在线的预测网络服务器,便于识别嗜热蛋白。在嗜热蛋白分析和进一步的实验研究中,这个预测将成为一个很有用的工具,此外,在这项研究中提出的方法可以推广到其他蛋白质的预测中。
为了能够得到更高精度的预测模型,接下来需要从以下方面来进行工作:
(1)实时更新搜索数据集,完善扩大数据集。比如可以将数据集筛选标准的最适温度范围扩大。
(2)提取新特征。例如还可以提取氨基酸、三肽、四肽甚至多肽作为特征,或者选取不同的物化性质作为特征等,筛选后寻求最佳精度,提高预测模型精度。
(3)开发更加准确、快速的分类预测算法。比如可以将随机森林和支持向量机相结合等。
(4)拓展研究。可以将蛋白质的热稳定性理论与其他生物学过程相结合进行研究,例如可以研究蛋白质的亚细胞定位与其耐热性的关系、嗜热菌在生物催化中的应用等相关领域。
References)
[1]LIANG H K, HUANG C M, KO M T, et al. Amino acid coupling patterns in thermophilic proteins[J]. Proteins,2005,59 (1): 58-63. DOI: 10.1002/prot.20386.
[2]ZHANG G Y, FANG B S. Application of amino acid distribution along the sequence for discriminating mesophilic and thermophilic proteins[J]. Process Biochemistry,2006,41(8): 1792-1798. DOI: 10.1016/j.procbio.2006.03.026.
[3]GROMIHA M M, SURESH M X. Discrimination of mesophilic and thermophilic proteins using machine learning algorithms[J]. Proteins,2008, 70(4): 1274-1279. DOI: 10.1002/prot.21616.
[4]MONTANUCCI L, FARISELLI P, MARTELLI P L, et al. Predicting protein thermostability changes from sequence upon multiple mutations[J]. Bioinformatics,2008,24(13): 190-195. DOI:10.1093/bioinformatics/btn166.
[5]WU L C, LEE J X, HUANG H D, et al. An expert system to predict protein thermostability using decision tree[J]. Expert Systems with Applications,2009, 36(5):9007-9014.DOI: 10.1016/j.eswa.2008.12.020.
[6]LI W Z, GODZIK A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences[J]. Bioinformatics, 2006,22(13):1658-1659. DOI: 10.1093/bioinformatics/btl158.
[7]CHOU K C. Prediction of protein cellular attributes using pseudo-amino acid composition[J]. Proteins, 2001,43(3):246-255.DOI: 10.1002/prot.1035.
[8]BHASIN M, RAGHAVA G P. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST[J]. Nucleic Acids Research,2004, 32(Web Server issue): W414-W419. DOI: 10.1093/nar/gkh350.
[9]CHOU K C. Some remarks on protein attribute prediction and pseudo amino acid composition[J]. Journal of Theoretical Biology, 2011,273(1): 236-247. DOI: 10.1016/j.jtbi.2010.12.024.
[10]TANG H, CHEN W,LIN H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique[J]. Molecular BioSystems, 2016,12(4):1269-275.DOI:10.1039/C5MB00883B.
[11]GOTTFRIES J, ERIKSSON L. Extensions to amino acid description[J]. Molecular Diversity ,2010, 14(4):709-718. DOI: 10.1007/s11030-009-9204-2.
[12]LUO L F. The degeneracy rule of genetic code[J]. Origins of Life and Evolution of Biospheres,1988,18(1-2): 65-70. DOI:10.1007/BF01808781.
[13]丁彦蕊,蔡宇杰,孙俊,等.基于SVM 和KNN 的蛋白质耐热性分类[J] 计算机工程与应用,2007,43(16):228-237.
DING Yanrui,CAI Yujjie,SUN Jun,et al. Protein heat tolerance classification based on SVM and KNN[J]. Computer Engineering and Applications,2007, 43(16):228-237.
[14]贾富仓,李华.基于随机森林的多谱磁共振图像分割[J]. 计算机工程, 2005,31(10): 159-161.
JIA Fucang,LI Hua. Multi spectral magnetic resonance image segmentation based on random forest[J]. Computer Engineering, 2005,31(10): 159-161.
[15]张光亚,方柏山. 基于氨基酸组成分布的嗜热和嗜冷蛋白随机森林分类模型[J].生物工程学报, 2008,24(2):302-308.
ZHANG Guangya, FANG Baishan. Based on the distribution of the amino acid composition is addicted to heat and psychrophilic protein random forest classification model[J]. Chinese Journal of Biotechnology,2008,24(2):302-308.
Prediction of thermophilic proteins based on physicochemical properties
DAO Fuying,CHEN Xinxin,LIN Hao*
(KeyLaboratoryforNeuro-InformationofMinistryofEducation,CenterforInformationalBiology,SchoolofLifeScienceandTechnology,UniversityofElectronicScienceandTechnologyofChina,Chengdu610054,China)
Thermophilic proteins can keep stability and activity at high temperature, which are ideal materials to study stability of proteins. Developing a valuable method to identify thermostability of protein would be helpful for protein engineering. In the present study, amino acid composition and physicochemical properties of protein have been thought of being related to the thermostability of protein. A reliable benchmark dataset including 915 thermophilic proteins and 793 non-thermophilic proteins is constructed for training and testing the proposed model in this article. We define protein samples using physicochemical properties and component of amino acid, so we design a descriptor which will combine dipeptide composition with nine physiochemical properties of amino acids. The results by support vector machine (SVM) with 5-fold cross-validation show that the best accuracy is 92.74% by using 290 features when the parameter gap is 0, indicating that our model holds very high potential to become a useful tool for the research on protein thermostability.
Thermophilic proteins;Thermostability;Pseudo amino acid composition;Physico-chemical roperties
2016-06-26;
2016-07-20.
四川省应用基础研究项目(2015JY0100);中央高校基本业务费(ZYGX2015J144,ZYGX2015Z006)。
刀福英,女,硕士研究生,研究方向:生物信息学;E-mail:18200234053@163.com.
*通信作者:林昊,男,研究员,硕士生导师,研究方向:生物信息学;E-mail:hlin@uestc.edu.cn.
10.3969/j.issn.1672-5565.2017.01.201606001
Q51
A
1672-5565(2017)01-001-06
猜你喜欢
海洋通报(2022年6期)2023-01-07 02:49:22
海南大学学报(自然科学版)(2020年2期)2020-07-21 10:56:02
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
中成药(2018年1期)2018-02-02 07:20:05
分析化学(2017年12期)2017-12-25 12:24:55
发酵科技通讯(2016年4期)2016-12-21 08:52:59
戏剧艺术(上海戏剧学院学报)(2016年6期)2016-01-03 08:22:36
动物医学进展(2015年10期)2015-12-07 05:46:19
学习月刊(2015年16期)2015-07-09 05:39:16
食品工业科技(2014年7期)2014-03-11 18:15:07
