
长野芽孢杆菌普鲁兰酶的同源建模及三维结构分析
2017-04-10 06:13:35韦旭钦李晓明廖东庆黄日波
生物信息学 2017年1期
韦旭钦,李晓明,廖东庆,黄日波
(1. 南宁邦尔克生物技术有限责任公司,南宁 530003;2. 广西大学生命科学与技术学院,南宁 530004;3. 广西科学院,南宁 530007)
长野芽孢杆菌普鲁兰酶的同源建模及三维结构分析
韦旭钦1*,李晓明1,廖东庆1,黄日波2,3
(1. 南宁邦尔克生物技术有限责任公司,南宁 530003;2. 广西大学生命科学与技术学院,南宁 530004;3. 广西科学院,南宁 530007)
普鲁兰酶(Pullulanase)是脱支酶,因其能水解葡聚糖的α-1,6-糖苷键而有不同的工业应用潜力。本研究通过同源建模和分子对接的方法对长野芽孢杆菌(Bacillusnaganoensis) 普鲁兰酶进行建模及其三维结构分析,表明该酶由CBM41-X45a-X25-X45b-CBM48-GH13_14多结构域组成,酶蛋白中心形成其催化区,催化区的Asp619、Glu648和 Asp733三个残基构成酶的催化三联体。同时,通过柔性对接研究了酶与底物分子相互作用的关系,并预测构成酶的活性中心相关氨基酸残基,为进一步改良酶的特性提供重要的理论依据。
长野芽孢杆菌;普鲁兰酶;同源建模;结构分析
普鲁兰酶(Pullulanase,EC 3.2.1.41)是一类水解α-1,6-糖苷键脱支酶,因其能专一性水解普鲁兰糖而得名,该酶可以水解支链淀粉的α-1,6-糖苷键而将淀粉分解为葡萄糖、麦芽糖、果糖等食用甜味剂;在以纤维素和淀粉质为原料的生物加工过程中,葡萄糖淀粉酶类水解直链的α-1,4-糖苷键,普鲁兰酶专门水解支链α-1,6-糖苷键,使支链淀粉型多糖的分支链脱离主链,形成一系列链长短不一的直链淀粉,可见普鲁兰酶和葡萄糖淀粉酶配合使用,可以显著提高淀粉类原料的利用率,因此,普鲁兰酶在食品、纺织和生物能源等领域有重要的用途[1]。
不同来源的普鲁兰酶由于其活性低和稳定性差等特点,限制了该类酶的工业化应用。研究发现,长野芽孢杆菌普鲁兰酶(Bacillusnaganoensispullulanase,BnPulB)的最适反应温度为62.5 ℃,在pH为4.5~5.0范围内保持90%的酶活性,这些酶学特性与淀粉糖化过程较高的温度(55~65 ℃)和微酸性环境(pH 4.5~5.5)相符合,表明长野芽孢杆菌普鲁兰酶有更大的工业应用潜力[2]。
同源建模法(Homology model)是从蛋白质的氨基酸序列出发预测其三维模型的常用方法,为研究未知结构蛋白质的功能提供基础,如果目的蛋白与模板蛋白的序列同一性大于60%,同源建模的结果将接近实验测试的结果[3]。为了进一步提高酶的活性和稳定性等特性,本研究通过对长野芽孢杆菌普鲁兰酶进行同源建模及其结构分析,并预测其活性部位和功能位点,为改良酶的性能提供理论参考。
1 材料与方法
1.1 序列来源
长野芽孢杆菌普鲁兰酶BnPulB的蛋白质序列来源于NCBI,该酶是由926个氨基酸残基组成的多肽,其GenBank登陆号为AEV53626,本研究所采用的6个普鲁兰酶序列及其晶体结构数据(见表1)取自蛋白质数据库(Protein Data Bank, PDB)[4],序列分析利用DiAlige服务器(http://www.genomatix.de/)进行[5]。
1.2 同源建模与结构分析
将BnPulB蛋白质序列提交到在线服务器SWISS-MODEL(http://swissmodel.expasy.org/)进行自动模式搜索和同源建模[6],用PyMol生物软件对其模型进行结构分析和作图。
1.3底物对接和活性位点分析
用Autodock 4.2生物软件对普鲁兰酶BnPulB与底物进行柔性对接及其活性位点分析[7]。
2 结果与分析
2.1BnPulB的同源建模
通过在线服务器SWISS-MODEL自动模式搜索(各参数为默认值),发现BnPulB与普鲁兰酶家族的有不同程度的序列同一性(见表1,各酶的PDB编号分别为2WAN、3WDI、2E9B、2YA0、2YOC和2FHC,aa表示蛋白序列的氨基酸残基个数),其中它与嗜酸普鲁兰芽孢杆菌普鲁兰酶(Bacillusacidopullulyticuspullulanase,BaPul13A)蛋白质序列同一性达64%,以BaPul13A三维结构(PDB:2WAN,1.65Å)为模板[8],对BnPulB进行同源建模,得到N端和C端分别缺失108和8个氨基酸的并由其Pro109~Gln918区间氨基酸肽段组成的分子量更小的结构,其QMEAN值仅为-0.01,加上BnPulB蛋白质序列与模板BaPul13A的同一性很高,可见BnPulB同源建模的结果接近实验测试的结果。

表1 BnPulB与6个有晶体结构的普鲁兰酶序列同一性Table 1 Pairwise sequence identity of BnPulB and 6 known crystal structure pullulanases %
2.2 BnPulB结构分析
将BnPulB模型与BaPul13A的进行整体结构比对分析,发现这两个酶各残基的Ca原子空间坐标几乎完全重叠,其总体RMSD 值仅为0.050Å,表明BnPulB结构与BaPul13A的结构非常相似,两者的整体结构几乎重叠在一起(见图 1)。

图 1 普鲁兰酶BnPulB与BaPul13A结构重叠分析*Fig.1 Structural superposition of pullulanase BnPulB and BaPul13A
*注:图中BnPulB的结构为蓝色,BaPul13A 结构(PDB编号:2WAN)为粉红色,BaPul13A催化三联体为Asp622、Glu651 和 Asp736三个氨基酸残基,对应BnPulB的分别为Asp619、Glu648 和 Asp733.
彩图见电子版(http://swxxx.alljournals.cn/ch/index.aspx)(2017年第1期DOI:10.3969/j.issn.1672-5565.2017.01.201606002)。
将BnPulB模型与表1中所述的6种普鲁兰酶同时进行结构比对(见图2),由于它们的序列长度和序列本身存在较大差异(见表1),它们的整体结构并不完全重叠(见图2(a)),但它们的活性中心的结构(底物结合口袋)却非常相似(见图2(b)),其氨基酸残基高度保守,都由空间坐标几乎重叠的Asp、Glu和Asp三个残基构成催化三联体(见图2(c)),表明不同来源的普鲁兰酶在漫长的家族进化过程中存在未知的可变性,它们整体的序列同一性可能很低,但其催化区是高度保守的,符合蛋白质生物进化的基本规律,可见长野芽孢杆菌普鲁兰酶的催化区同样具有普鲁兰酶家族共同的结构特点。
已有研究表明,BaPul13A是由Asp622、Glu651和Asp736三个氨基酸残基构成其催化三联体,在CAZY GH13糖苷酶家族中,这三个位于催化区的氨基酸残基非常保守,在酶的催化过程中,Asp622充当进攻底物的亲核基团,Glu651作为广义的酸碱催化剂,Asp736对酶-底物复合物的过度态起到稳定性的作用[8]。通过结构比对分析,可以预测,构成对应的长野芽孢杆菌BnPulB催化三联体是由Asp619、Glu648和Asp733三个氨基酸残基构成(如图1和图2(c)所示),这三个残基与表1所述的6种同类普鲁兰酶催化三联体的空间坐标大致重叠(见图2(c)),因此这三个残基在酶的催化过程中可能起到与BaPul13A催化三联体相似的关键作用。

图 2 BnPulB与6种普鲁兰酶的结构比对分析*Fig.2 Structural superposition of BnPulB with 6 known pullulanases
注:图中BnPulB (蓝色)、2WAN(粉红色)、3WDI (红色)、2E9B (青色)、2YA0 (黄色)、2YOC (红色)、2FHC (灰色)。
彩图见电子版(http://swxxx.alljournals.cn/ch/index.aspx)(2017年第1期DOI:10.3969/j.issn.1672-5565.2017.01.201606002)。
BaPul13A是由921个氨基酸残基组成CBM41-X45a-X25-X45b-CBM48-GH13_14多结构域的蛋白,其中X45a-X25-X45b 的功能未知,CBM48为糖原结合结构域,催化结构域GH13_14属于 CAZY GH13家族,其N端1~100氨基酸为功能未明的结构域CBM41,CBM41高度无序而无法模拟其结构[7]。Chen等[9]最新研究表明,删除BaPul13A酶的CBM41结构域,能有效地提高该酶的可溶表达和分泌水平,突变酶的活力是野生酶的2.9倍,并且其作用的温度和pH与野生酶的相当,由此可见CBM41结构域是酶分子上冗余的元件,删除该结构域的突变酶更适合工业化应用。因此,可以推断,长野芽孢杆菌普鲁兰酶BnPulB对应的N端1~108氨基酸可能是酶分子上冗余的序列,删除这个结构可能得到分子量更小的结构更稳定的突变酶。
Wang等[10]近期研究发现,删除长野芽孢杆菌BnPulB酶N端前106个氨基酸残基,或C端9个,或C端36个残基的突变体,突变酶的催化温度和pH没有改变,突变酶对底物亲和力和催化效率有不同程度的改观,与本研究预测的结果基本相符。
通过结构比对分析,BnPulB各结构域在酶分子上的氨基酸区间分布如图3(a)所示,各结构域在酶空间结构的分布如图3(b)所示,其中X45a、X25和X45b结构域远离酶的催化区(GH13_14结构域),它们可能有助于结合更大分子的底物,与酶的结构稳定性有关;CBM48是糖原结构域,它和催化区的GH13_14结构域邻近,这两个结构域的结合维持了这个脱支酶的基本功能。BnPulB的催化三联体位于GH13_14结构域,底物位于该结构域的深沟中,GH13_14结构域与酶的催化作用密切相关,改变这个结构域的相关氨基酸可能会明显影响酶的活性和催化稳定性。Chen等[11]将BaPul13A酶位于GH13_14结构域的3个氨基酸残基同时突变(E518I-S662R-Q706P),突变体酶在60 ℃的半衰期是野生型的11倍,最适反应温度也从60 ℃提升到65 ℃。由于与BaPul13A酶的空间结构非常相似,若将BnPulB空间位置对应的氨基酸残基(E515、A659和L703)进行相应的突变,也可能得到相应性能更稳定的突变酶。这种通过比对结构类似的(关键位点的氨基酸残基存在差别),并经过突变其相应关键位点氨基酸残基来影响酶功能的方法(类比差别法),将为酶的分子改良开辟一条新的途径。
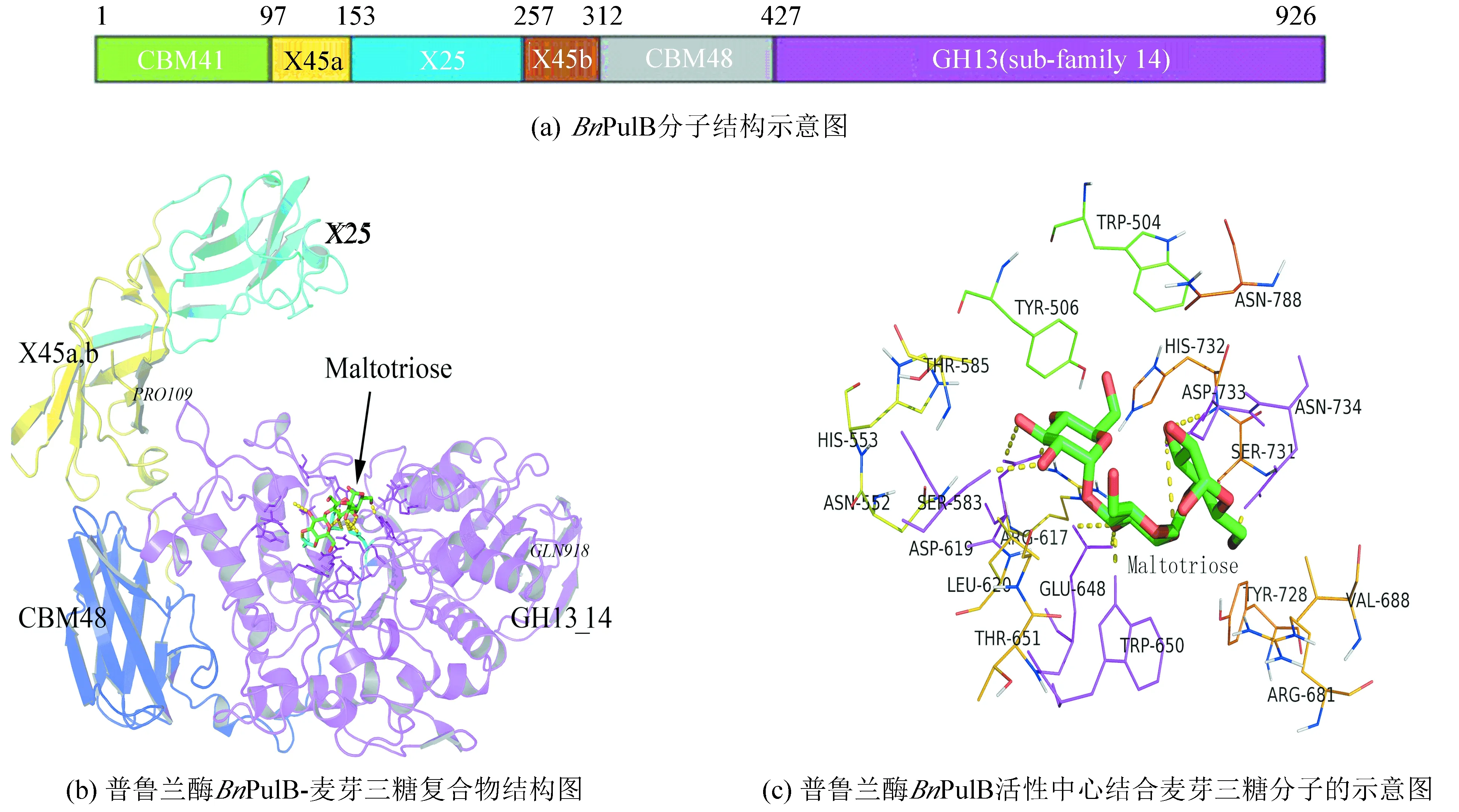
图 3 长野芽孢杆菌普鲁兰酶(BnPulB)的三维结构图*Fig.3 3-D structure of the Bacillus naganoensis pullulanase(BnPulB)
注:图(b)的BnPulB结构用卡通图形显示,结构域X45为黄色,X25的为青色,CBM48为蓝色,GH13_14的为粉红色,麦芽三糖分子为棍棒模型;图(c)活性中心邻近氨基酸残基以绳形显示,麦芽三糖(棍棒模型)与氨基酸残基(粉红色)形成的氢键以虚线显示。
*彩图见电子版(http://swxxx.alljournals.cn/ch/index.aspx)(2017年第1期DOI:10.3969/j.issn.1672-5565.2017.01.201606002)。
2.3底物对接
用软件Autodock 4.2将底物与酶分子进行对接,将BnPulB的催化三联体Asp619、Glu648和Asp733设定为柔性残基,经分析得到这三个氨基酸残基的中心坐标,将Gridbox中心设定为X:39.21,Y:37.82,Z:-29.32,将Gridbox大小设定为30×52×32(单位Å),格点间距为默认值0.375 Å,对接过程选择Lamarckian genetic algorithm (LGA)遗传算法,其他条件均为默认值。由于普鲁兰糖是由α-1,4-糖苷键连接的麦芽三糖重复单位经α-1,6-糖苷键聚合而成的分子量达2 万~200 万之间直链状多糖,本研究以构成普鲁兰糖单元的麦芽三糖为配体,麦芽三糖分子从厌氧芽孢杆菌 LM18-11(Anoxybacillussp. LM18-11)的同类普鲁兰酶晶体结构(PDB:3WDI)中提取[12]。
分子对接结果见图3(b),底物分子深埋在酶分子的GH13_14结构域空穴中,底物结合口袋由多个氨基酸残基组成,其结合自由能为-2.75 kcal/mol,抑制剂常数Ki=9.58 mM,底物分子的成功对接,为研究酶与底物的相互作用,研究酶的活性中心及其分子改良奠定了理论基础。
2.4 活性中心分析
根据底物对接的结果,表明构成BnPulB的底物结合区和催化区的结构非常复杂,如图3(c)所示,由Trp504、Try506、Asn552、His553、Ser583、Thr585、Arg617、Asp619、Leu620、Glu648、Trp650、Thr651、Arg681、Val688、Tyr728、Ser731、His732、Asp733、Asn734和Asn788 20个氨基酸残基组成酶的活性中心,其中催化区直接与麦芽三糖形成的氢键的有Ser583、Asp619、Glu648、Trp650、Asp733和Asn734 6个关键氨基酸残基,其催化三联体由Asp619、Glu648和Asp733 3个氨基酸残基组成,这3个残基在同类酶中高度保守(见图2(c)),直接突变这些位点,可能使酶失去活性,因此,可以考虑对构成其活性中心的其他非形成氢键的残基着手,进行适当的突变来优化其底物结合口袋和催化区的结构,从而提高酶的催化效率;也可以通过研究与活性中心邻近(5~15 Å)的氨基酸残基来优化酶的构象或微环境来改善酶的特性[13]。
3 结论与讨论
本研究对长野芽孢杆菌普鲁兰酶BnPulB进行同源建模,构建了BnPulB蛋白的三维结构。通过理论模拟表明,该普鲁兰酶具有CBM41-X45a-X25-X45b-CBM48-GH13_14多结构域的结构,其催化区位于GH13_14结构域,它的催化三联体由Asp619、Glu648和Asp733三个氨基酸残基组成,符合已知同类普鲁兰酶结构的相关特性;酶与底物对接及其活性中心的预测和分析表明,普鲁兰酶家族的底物结合口袋非常相似,BnPulB与底物形成氢键的氨基酸残基为Ser583、Asp619、Glu648、Trp650、Asp733和Asn734,突变这些残基可能使酶失去活性,所以在分子改良时,可考虑对BnPulB活性中心没有形成氢键的残基着手来改良酶的特性。本文的研究方法具有操作简单和直观易懂等特点,理论成果对酶的分子改良有一定的参考意义,其不足之处是在同类酶非保守区或非同源性蛋白的预测可能存在较大偏差,其研究方法和理论成果还有待更多的实验验证。
References)
[1]GUZMAN-MALDONADO H, PAREDES-LOPEZ O. Amylolytic enzymes and products derived from starch: A review [J].Critical Reviews in Food Science and Nutrition,1995,35(5): 373-403.DOI: 10.1080/10408399509527706.
[2]严伟, 聂尧, 徐岩. 长野芽孢杆菌(Bacillusnaganoensis)普鲁兰酶在大肠杆菌中的活性表达与分泌调控 [J]. 微生物学报,2013,53(2): 145-153.
YAN Wei,NIE Yao,XU Yan. Expression and secretion regulation ofBacillusnaganoensispullulanase in recombinantEscherichiacoli[J].Acta Microbiologica Sinica,2013,53(2): 145-153.
[3]KACZANOWSKI S, ZIELENKIEWICZ P. Why similar protein sequences encode similar three-dimensional structures? [J].Theoretical Chemistry Accounts,2010(125): 543-550.DOI: 10.1007/s00214-009-0656-3.
[4]BERMAN H M, WESTBROOK J, FENG Z, et al. The protein data bank [J]. Nucleic Acids Research,2000,28(1): 235-242.
[5]MORGENSTERN B, FRECH K, DRESS A, et al. Dialign: Finding local similarities by multiple sequence alignment [J].Bioinformatics,1998,14(3): 290-294.DOI: 10.1093/bioinformatics/14.3.290.
[6]SCHWEDE T, KOPP J, GUEX N, et al. Swiss-model: An automated protein homology-modeling server [J].Nucleic Acids Research,2003,31(13): 3381-3385.DOI: 10.1093/nar/gkg520.
[7]MORRIS G M, HUEY R, LINDSTROM W, et al. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility [J].Journal of Computational Chemistry,2009,30(16): 2785-2791.DOI: 10.1002/jcc.21256.
[8]TURKENBURG J P, BRZOZOWSKI A M, SVENDSEN A, et al. Structure of a pullulanase fromBacillusacidopullulyticus[J].Proteins Structure Function &Bioinformatics,2009,76(2): 516-519.DOI: 10.1002/prot.22416.
[9]CHEN A N, SUN Y, ZHANG W, et al. Downsizing a pullulanase to a small molecule with improved soluble expression and secretion efficiency inEscherichiacoli[J].Microbial Cell Factories,2016,15(9):1-10.DOI: 10.1186/s12934-015-0403-5.
[10]WANG X Y, NIE Y, MU X Q, et al. Disorder prediction-based construct optimization improves activity and catalytic efficiency ofBacillusnaganoensispullulanase [J].Scientific Reports,2016,6(24574):1-9.DOI: 10.1038/srep24574.
[11]CHEN A N, LI Y M, NIE J Q, et al. Protein engineering ofBacillusacidopullulyticuspullulanase for enhanced thermostability using in silico data driven rational design methods [J].Enzyme and Microbial Technology,2015(78): 74-83.DOI: 10.1016/j.enzmictec.2015.06.013.
[12]XU J Y, REN F F, HUANG C H, et al. Functional and structural studies of pullulanase fromAnoxybacillussp. LM18-11[J]. Proteins Structure Function &Bioinformatics,2014,82(9): 1685-1693.DOI: 10.1002/prot.24498.
[13]MORLEY K L, KAZLAUSKAS R J. Improving enzyme properties: When are closer mutations better? [J]. Trends in Biotechnology,2005,23(5): 231-237.DOI: 10.1016/j.tibtech.2005.03.005.
Homology modeling and three-dimensional structure analysis ofBacillusnaganoensispullulanase
WEI Xuqin1*, LI Xiaoming1, LIAO Dongqing1, HUANG Ribo2,3
(1.GuangxiNanningBiocloneBiotechnologyCo.,Ltd.,Nanning530003,China;2.CollegeofLifeScienceandTechnology,GuangxiUniversity,Nanning530004,China;3.GuangxiAcademyofSciences,Nanning530007,China)
Pullulanase is a debranching enzyme that specifically hydrolyzes the α-1,6-glycosidic bonds in complex carbohydrates and has great potential in various industries. Here, we investigate the structural characteristics of theBacillusnaganoensispullulanase (BnPulB) by homology modeling and molecule docking. Results show that theBnPulB structure comprises the CBM41-X45a-X25-X45b-CBM48-GH13_14 multi-domain architecture and the central region of the protein forms the catalytic domain, and the highly conserved Asp619, Glu648, and Asp733 residues are identified to be catalytic triad. In addition, flexible docking studies of the enzyme-substrate system show the interactions betweenBnPulB and its substrate, maltotriose, and some conserved residues locate in the active centre that participate in ligand binding site are predicted. This study may provide important information for the design of new pullulanase with novel properties.
Bacillusnaganoensis;Pullulanase; Homology modeling; Structure analysis
2016-06-18 ;
2016-09-08.
南宁市科技型中小企业技术创新基金资助项目 (20146371)。
10.3969/j.issn.1672-5565.2017.01.201606002
Q514
A
1672-5565(2017)01-027-06
*通信作者:韦旭钦,男,理学博士,高级工程师,研究方向:微生物发酵与分子酶工程;E-mail:xqweigxu@163.com.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
食品与药品(2020年1期)2020-03-10 03:25:00
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
居业(2014年4期)2014-06-06 21:15:05
食品工业科技(2014年13期)2014-03-11 18:16:48
中国粮油学报(2014年8期)2014-02-06 01:34:10
生物加工过程(2013年1期)2013-10-25 06:25:00
知识窗(2013年3期)2013-05-14 09:08:05
