
病例-对照设计的似然比罕见变异关联性检验的构建和模拟评价*
2017-04-08 03:24:44金英良张耀东
郑州大学学报(医学版) 2017年2期
曾 平,赵 杨,陈 峰,金英良,张耀东
1)徐州医科大学公共卫生学院流行病与卫生统计学教研室 江苏徐州 221004 2)南京医科大学公共卫生学院生物统计学系 南京 210009
病例-对照设计的似然比罕见变异关联性检验的构建和模拟评价*
曾 平1)△,赵 杨2),陈 峰2),金英良1),张耀东1)
1)徐州医科大学公共卫生学院流行病与卫生统计学教研室 江苏徐州 221004 2)南京医科大学公共卫生学院生物统计学系 南京 210009
△男,1982年7月生,博士,讲师,研究方向:高维数据分析和统计遗传学方法研究,E-mail:zpstat@xzhmu.edu.cn
罕见变异;关联性分析;logistic混合模型;似然比检验
目的:发展适用于病例-对照设计的似然比罕见变异关联性分析方法。方法:在logistic混合模型的框架下基于PQL算法建立伪数据,将二分类表型转化为连续型表型的关联性分析,然后借助线性混合模型的方差成分检验执行关联性分析。采用Monte Carlo模拟评价该方法的有效性,并与现有方法进行对比。结果:模拟显示,在不同情况下包括似然比检验在内的所有统计检验都能有效控制Ⅰ型错误;在效应方向相同情况下,Burden检验、SKAT-O和MiST的统计效能高;在效应方向不同的情况下似然比检验优于其他方法。结论:基于logistic混合模型和PQL算法的似然比检验可有效用于病例-对照设计的罕见变异关联性分析。
过去十年,尽管全基因组关联研究取得了巨大成功,但对许多复杂疾病而言,已发现的绝大部分常见位点效应都很弱,仅能解释极少一部分遗传度,未能取得预期的突破性进展[1-3]。越来越多的证据表明罕见变异对复杂疾病有实质性的贡献[4],支持的论据包括[5]:①进化理论暗示与疾病相关的遗传变异应该是近期发生的并且是罕见的。②人群遗传研究的许多经验证据表明对人类健康有害的变异是罕见的。③一些心理疾病由罕见的拷贝数变异引起。④许多不常见的家族性疾病由罕见遗传位点变异所致。⑤由罕见变异引起的虚拟关联可解释部分常见变异与疾病的关联[6]。然而,由于最小等位基因频率极低,很难对罕见变异进行关联性研究。作者主要针对病例-对照设计,在广义混合效应模型框架下构建基于似然比检验的罕见变异关联性分析方法,通过Monte Carlo评价该方法的性质、表现。
1 方法原理和模拟设置
1.1 方法原理 设Y=1和0分别表示病例和对照,X为协变量,G为K维基因型,样本量为n。采用logistic混合效应模型描述Y、X和G之间的关系:
式中,W=Beta(MAFk|1,25),为K维的对角权重矩阵,Beta(a1,a2)为Beta函数,MAF表示最小等位基因频率,τ2为方差参数。检验G是否与Y存在关联等价于检验H0:τ2=0。采用惩罚拟似然算法(penalized quasi-likelihood,PQL)估计模型的参数,通过R软件中的函数glmmPQL[7]。具体而言,PQL算法在伪对数极大(ml)或限制性极大(reml)似然函数和工作变量Y′间迭代:

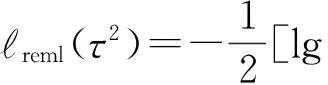
lg|X′D-1X|]
Y′=Xβ+Gb+ε
其中R是对角元素为μ(1-μ)的n维对角矩阵。当PQL算法收敛时,工作应变量为
则
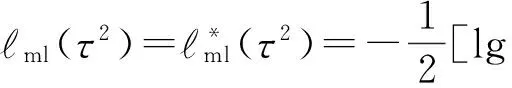

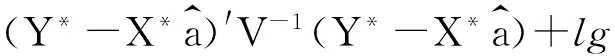
V=τ2G*WWG*′+In
这样就将病例-对照设计的方差成分检验转化成为了连续型表型的方差成分检验。建立(伪)似然比统计量
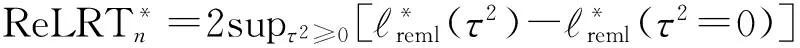
也即是,在执行方差成分检验时只要将伪数据当作真实数据即可。
1.2模拟设置 采用COSI软件模拟联合模型产生欧洲人群的罕见遗传变异基因型[8]。协变量分别为相互独立的标准正态随机变量(X1)和率为0.50的二项分布随机变量(X2)。Y从二项分布产生
其中,C为关联位点数,b为效应;Ⅰ型错误控制时b=0,统计效能评价时b=0.4|lgMAF|。Ⅰ型错误控制和统计效能模拟次数分别为10 000和5 000,检验水准α设定为0.01,Ⅰ型错误和统计效能由P≤α(=0.01)的比例估计。统计分析在R软件中完成,Burden检验、SKAT和SKAT-O执行软件包SKAT[9],MiST[10]和GenRF[11]均执行相应函数。分别考察固定基因片段和不同基因片段条件下的Ⅰ型错误控制和统计效能分析。
2 结果
2.1 模拟1:固定基因片段条件下的Ⅰ型错误控制和统计效能分析 选择30%的基因片段,样本量为400、600、800和1 000时Ⅰ型错误控制情况见表1。结果表明在固定基因片段和不同样本量条件下,SKAT和MiST展现出保守的倾向,而其他方法基本上都能较好地控制Ⅰ型错误。

表1 固定基因片段和不同样本量条件下的Ⅰ型错误控制
α=0.01。
选择30%的基因片段(罕见变异平均个数约70),将其中30%的罕见变异设为关联位点,并假设3种情况:①所有关联位点效应方向为正;②30%关联位点效应方向为负,70%效应方向为正;③50%关联位点效应方向为负,50%效应方向为正。样本量为400、600、800和1 000。为了解大样本情况下似然比检验的表现,可将样本量增加到2 000。结果显示,当所有关联位点效应方向都为正时,Burden检验、SKAT-O和MiST的统计效能更高,其次是LRT和ReLRT,而SKAT、GenRF的效能最小(表2)。当关联位点效应方向不一致时,LRT、ReLRT和SKAT表现出更高的效能,而GenRF、SKAT-O和MiST次之,Burden检验的效能最低(表3、4)。相对效应方向一致的情况,效应方向相反时所有检验方法的效能均降低,Burden检验、SKAT-O和MiST降低最明显,而SKAT、LRT和ReLRT降低较少,GenRF降低最少,LRT和ReLRT降低程度不如SKAT(表5、6);且正负向效应比例相差越小,效能损失越大;当效应方向一致时LRT和ReLRT不如其他方法,但是当效应方向异质时它们优于其他方法,效能损失相对较低。

表2 所有关联位点效应方向都为正时的统计效能

表3 30%关联位点效应方向都为负时的统计效能
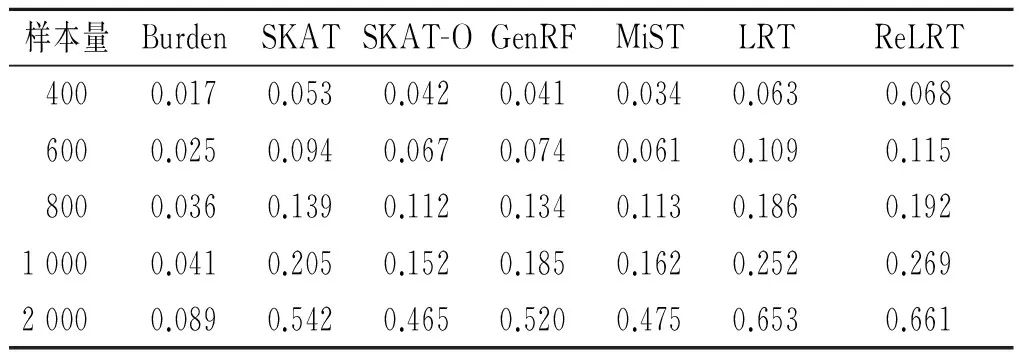
表4 50%关联位点效应方向都为负时的统计效能

表5 30%关联位点效应方向都为负时的统计效能相对所有关联位点效应方向都为正时的效能损失
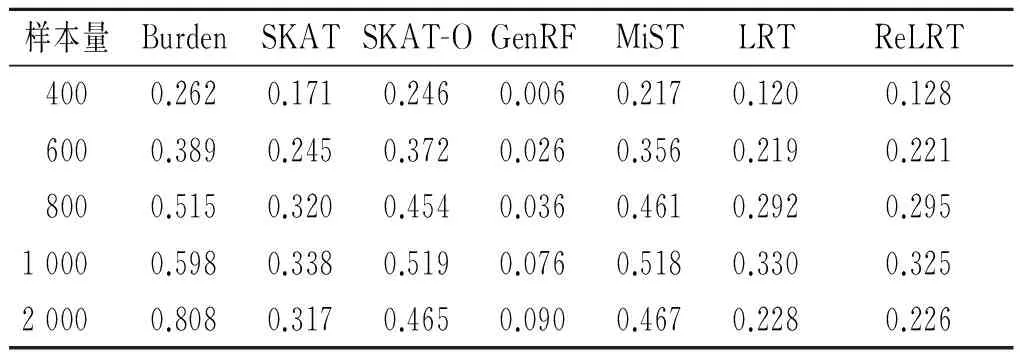
表6 50%关联位点效应方向都为负时的统计效能相对所有关联位点效应方向都为正时的效能损失
2.2 模拟2:可变基因片段条件下的Ⅰ型错误控制和统计效能分析 选择10%到90%的基因片段(对应人类基因组不同染色体基因上可能包含不同个数的罕见变异),样本量固定为1 000时Ⅰ型错误控制情况见表7。LRT和ReLRT在罕见变异位点数较少时能够很好控制Ⅰ型错误,但当位点数逐渐增大时,会产生比名义检验水准更高的Ⅰ型错误;而其他方法基本都能较好控制Ⅰ型错误。
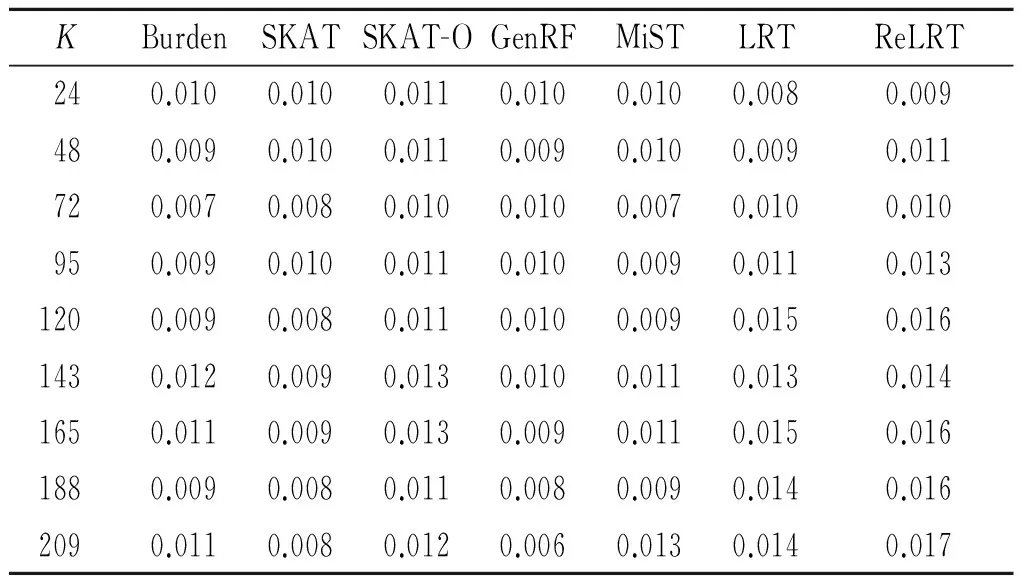
表7 可变基因片段和固定样本量条件下的Ⅰ型错误控制
K:包含在不同基因片段区域内的平均罕见变异个数。
选择10%到90%的基因片段,将其中30%的位点设为关联位点,样本量为1 000,假设50%的位点效应b=-0.4|lgMAF|,而剩下50%的位点效应b=0.4|lgMAF|。结果显示,随着罕见位点数的增加,每种检验方法的统计效能也相应增加,但在相同位点数情况下,因为假设正向和负向效应同时存在,因此LRT和ReLRT的效能最高,其次是MiST和SKAT,而Burden检验的效能最低(表8)。
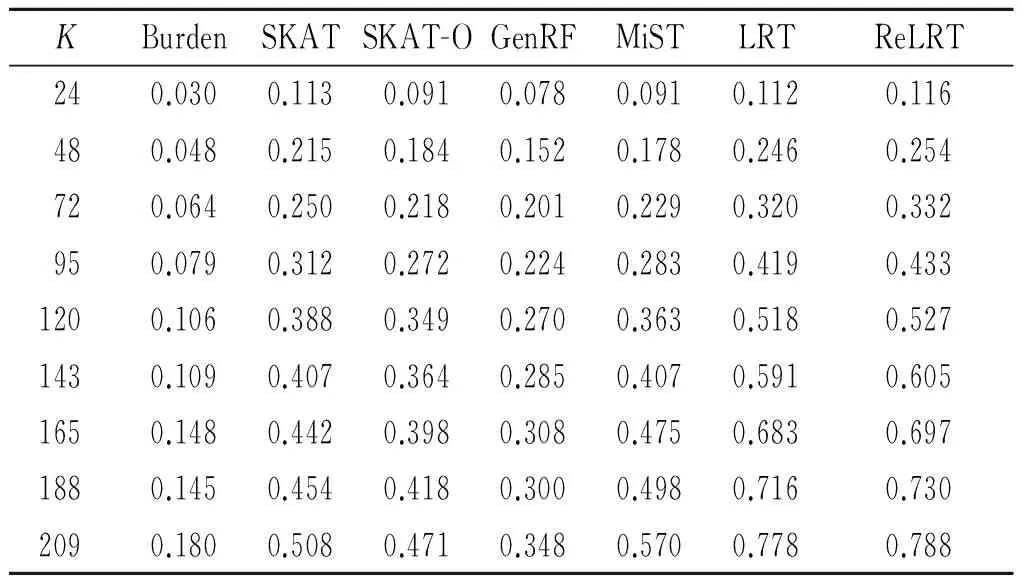
表8 可变基因片段条件下的统计效能
K:包含在该亚片段区域内的平均罕见变异位点数。
2.3 运算时间 假设检验1 000个基因,罕见变异位点数分别为10、30、50、80和100,样本量为2 000。计算在2.40 GHz和2.00 Gb内存个人电脑上的执行时间。表9显示,Burden检验的计算速度最快,SKAT、SKAT-O和MiST次之,LRT和ReLRT的速度较之更慢,GenRF最慢。

表9 运算时间 s
3 讨论
作者将病例-对照设计的罕见变异关联性分析转化为在广义混合效应模型框架下的似然比方差成分检验。虽然这种转化在概念上简单,但是存在很多难题,包括参数估计、对数似然值的计算和无效分布的推导。作者通过PQL算法估计参数,然后在工作应变量的基础上建立伪数据;将伪数据看作原始数据并且将工作应变量看作连续表型,这样问题就转化为了线性混合效应模型的方差成分检验。模拟结果表明,SKAT和MiST等方法在小样本时相对保守,而似然比检验能够很好地控制Ⅰ型错误,且具有更高的统计效能,但运算速度较慢,其计算主要消耗在估计logistic混合效应模型和模拟无效分布。
似然比检验依赖于PQL算法,而PQL算法事实上是一种有偏估计,尤其是在稀疏logistic回归中[12-15]。在稀疏情况下工作应变量的正态近似效果差,导致参数估计偏倚。但从模拟结果来看这种有偏性对Ⅰ型错误影响不大,尤其是在罕见变异位点数较少时。然而,罕见变异在位点数比较多时伪似然比检验会变得更加自由,产生比名义检验水准更高的Ⅰ型错误。其原因可能是:①点数增加意味着模型中随机效应的维数过大,此时PQL算法可能变得更加有偏。②构建的伪数据对正态分布的近似性较差。因此,有必要进一步研究似然比检验在参数无偏估计时的表现,以及提高其计算速度。
[1]MANOLIO A,COLLINS S,COX J,et al.Finding the missing heritability of complex diseases[J].Nature,2009,461(7265):747
[2]EICHLER EE,FLINT J,GIBSON G,et al.Missing heritability and strategies for finding the underlying causes of complex disease[J].Nat Rev Genet,2010,11(6):446
[3]MAHER B.Personal genomes: The case of the missing heritability[J].Nature,2008,456(7218):18
[4]BODMER W,BONILLA C.Common and rare variants in multifactorial susceptibility to common diseases[J].Nat Genet,2008,40(6):695
[5]GIBSON G.Rare and common variants: twenty arguments[J].Nat Rev Genet,2012,13(2):135
[6]DICKSON P,WANG K,KRANTZ I,et al.Rare variants create synthetic genome-wide associations[J].PLoS Biol,2010,8(1):e1000294
[7]VENABLES WN,RIPLEY BD.Modern applied statistics with S[M].4th ed.Heidelberg:Springer-Verlag,2002.
[8]SCHAFFNER SF,FOO C,GABRIEL S,et al.Calibrating a coalescent simulation of human genome sequence variation[J].Genome Res,2005,15(11):1576
[9]WU MC,LEE S,CAI T,et al.Rare-variant association testing for sequencing data with the sequence kernel association test[J].Am J Hum Genet,2011,89(1):82
[10]DUCHESNE P,DE MICHEAUX PL.Computing the distribution of quadratic forms: Further comparisons between the Liu-Tang-Zhang approximation and exact methods[J].Comput Stat Data Anal,2010,54(4):858
[11]HE ZH,ZHANG M,ZHAN XW,et al.Modeling and testing for joint association using a genetic random field model[J].Biometrics,2014,70(3):471
[12]BRESLOW NE,CLAYTON DG.Approximate inference in generalized linear mixed models[J].J Am Stat Assoc,1993,88(421):9
[13]LIN X,BRESLOW NE.Bias correction in generalized linear mixed models with multiple components of dispersion[J].J Am Stat Assoc,1996,91(435):1007
[14]LIN X.Variance component testing in generalised linear models with random effects[J].Biometrika,1997,84(2):309
[15]WANG N,LIN XH,GUTIERREZ RG,et al.Bias analysis and SIMEX approach in generalized linear mixed measurement error models[J].J Am Stat Assoc,1998,93(441):249
(2016-05-17收稿 责任编辑王 曼)
Development and simulations of likelihood ratio test for rare variants association analysis in case control studies
ZENGPing1),ZHAOYang2),CHENFeng2),JINYingliang1),ZHANGYaodong1)
1)DepartmentofEpidemiologyandBiostatistics,SchoolofPublicHealth,XuzhouMedicalUniversity,Xuzhou,Jiangsu221004 2)DepartmentofBiostatistics,SchoolofPublicHealth,NanjingMedicalUniversity,Nanjing210009
rare variant;association study;logistic mixed model;likelihood ratio test
Aim: To develop a likelihood ratio test for rare variant association analysis in case control studies. Methods: The likelihood ratio test was constructed under the framework of logistic mixed models; a new pseudo-data set was obtained via the working response in the PQL algorithm, which transformed the problem of testing rare variants association in case control studies into the problem of testing rare variants association with continuous under the framework of linear mixed models. The Monte Carlo simulation was conducted to evaluate the proposed test and to compare with existing methods.Results: The simulations showed that all the methods could control the type Ⅰ error correctly. When the effects of the causal rare variants were in the same direction, the Burden test, SKAT-O and MiST were most powerful; while if both positive and negative effects were present, the likelihood ratio test outperformed the others.Conclusion: The likelihood ratio test developed using the logistic mixed models and the PQL algorithm can be applicable to rare variant association analysis in case control studies.
10.13705/j.issn.1671-6825.2017.02.004
*国家自然科学基金项目 81402765;国家统计局全国统计科学研究项目 2014LY112
R195.1
猜你喜欢
内蒙古统计(2021年4期)2021-12-06 02:49:20
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
当代陕西(2019年15期)2019-09-02 01:52:00
测控技术(2018年4期)2018-11-25 09:46:52
学苑创造·A版(2018年11期)2018-02-01 06:29:20
上海精神医学(2017年5期)2017-11-29 06:03:10
读者(2017年5期)2017-02-15 18:04:18
百科知识(2015年18期)2015-09-10 07:22:44
统计与决策(2012年14期)2012-07-25 08:15:34
