
基于EST-SSR标记的贵州野生刺梨居群遗传多样性分析
2017-04-07 00:56张怀山鄢秀芹鲁敏王道平安华明
中国农业科学 2017年6期
张怀山,鄢秀芹,鲁敏,王道平,安华明
(1贵州大学农学院/贵州省果树工程技术研究中心,贵阳 550025;2贵州省中国科学院天然产物化学重点实验室,贵阳550002)
基于EST-SSR标记的贵州野生刺梨居群遗传多样性分析
张怀山1,鄢秀芹1,鲁敏1,王道平2,安华明1
(1贵州大学农学院/贵州省果树工程技术研究中心,贵阳 550025;2贵州省中国科学院天然产物化学重点实验室,贵阳550002)
【目的】通过分析评价贵州野生刺梨(Rosa roxburghii Tratt)资源居群遗传多样性和遗传结构,为刺梨资源的保护和发掘利用提供科学依据。【方法】从刺梨EST-SSR引物中筛选扩增效果好、多态性较高的10对引物,对收集的12个野生刺梨自然居群(共255份资源)的遗传多样性及遗传结构应用POPGENE 1.31软件进行分析,并根据Nei’s标准遗传距离,利用NTSYS-pc2.10e软件对各居群进行聚类分析,同时进行Nei’s遗传距离与地理距离的Mantel相关性检验。【结果】居群内Shannon信息指数(I)为0.62—0.88,基因多样性指数(Nei’s)为0.40—0.52,且除福泉(FQ)和晴隆(QL)居群外,其余10个居群的多态位点百分率均为100%。此外,卡方检验显示12个居群在大多数位点上等位基因显著偏离Hardy-Weinberg平衡(P<0.05),且居群内近交系数(Fit)、总近交系数(Fis)均为负值,居群间分化系数Fst平均值0.0403,居群间基因流Nem平均值为5.9484、遗传一致度GI为0.9068—0.9926、最大Nei’s遗传距离为0.0978。各自然居群间Nei’s遗传距离与地理距离存在显著相关性(r=0.2498,P=0.9512)。【结论】10对EST-SSR引物具有高度的多态性,可作为有效的遗传标记用于刺梨居群遗传多样性和遗传结构评价。贵州省野生刺梨自然居群内的遗传多样性较高,存在杂合度过剩的现象,且绝大多数的遗传变异发生在居群内;居群间具有基因交流频繁、遗传一致度高、Nei’s遗传距离小等特点。
刺梨;SSR;遗传多样性;居群遗传结构
0 引言
【研究意义】刺梨(Rosa roxburghii Tratt,2n=2x=14)为蔷薇科(Rosaceae)蔷薇属(Rosa)植物,是中国特有的果树资源。因其果实含有丰富的营养物质和药用保健成分,尤其是极高的维生素C而倍受关注。刺梨作为贵州省重点发展的特色产业,种植面积已超过 300 km2。同时,贵州省还含有丰富的野生刺梨资源,多分布于海拔1 000—1 600 m的山区、丘陵地带[1-2]。这些丰富的野生资源无疑是刺梨品种选育和产业可持续发展的重要基础和必要前提。然而,近年来对野生刺梨资源进行调查收集时发现,贵州野生刺梨分布面积和群体数量正在急剧减少,这使得开展刺梨资源的收集、遗传多样性评价、保护性开发等方面的工作势在必行。【前人研究进展】遗传多样性,包括群体遗传结构的研究,有利于对种质资源提出科学合理的保护策略,尤其是对于现今正遭受人类直接干扰和破坏的物种。SSR标记是研究生物遗传多样性的重要方法,已在蔷薇科植物群体遗传学、种质资源、分类学与种系发生学等方面得到广泛应用。ZHANG等[3]利用8对SSR标记对中国新疆伊犁地区4个种下居群109个新疆野苹果实生株系,进行群体遗传结构研究,结果表明巩留县遗传多样性最为丰富,故在制定原位种质保护计划时应优先考虑巩留县居群。ZONG等[4]利用14对SSR标记分析了川梨的遗传多样性和遗传分化,这14个位点在幼苗种群中显示较高的多态性,并确定出3个群体将成为重点保护和调查使用的对象。LIU等[5]利用14对基因组SSR引物研究了中国浙江省8个居群共77份豆梨资源的遗传多样性和种群结构,结果显示地理距离是目前豆梨遗传结构形成的一个关键因素,并确定FY群体是最适合进行原地保护的种群。陈娇等[6]采用10对SSR引物对四川野生中国樱桃5个居群共133株的遗传多样性水平及居群遗传结构进行了研究,并提出野生中国樱桃的保护利用策略。MENG等[7]利用SSR标记及单拷贝核基因对云南特有物种大花香水月季的遗传结构进行了研究,为保护这一濒危物种提供了重要的规划和决策。然而,刺梨种质资源分子水平的研究起步较晚,迄今为止,相关研究仅采用了少数几个刺梨材料作为参比试材研究蔷薇属不同种间亲缘进化的关系[8-13],以及开展了刺梨种内遗传多样性研究,但仅局限于30个刺梨样品的RAPD分析[14-15]。此外,笔者课题组在开发EST-SSR标记的过程中,曾涉及16份刺梨资源作为研究材料[16-17]。由此可知,刺梨种质资源在分子水平上的遗传多样性研究基础还相当薄弱,表现为样品数量不足、代表性差,且缺乏群体遗传结构的研究。【本研究切入点】SSR标记根据其来源可分为基因组SSR和EST-SSR,目前对刺梨遗传背景的研究刚刚起步[18],基因组信息较缺乏,可用的遗传标记较少,EST-SSR作为一种新型的分子标记,不仅具有传统SSR标记多态性高、共显性与重复性好等特点,更重要的是开发成本低;而且由于EST-SSR来源于表达的基因组区域,可直接反映相关基因的多样性,在不同物种间也具有良好的通用性[19]。本研究基于课题组前期对刺梨果实转录组测序获得的数据,筛选有效的刺梨EST-SSR引物,以研究刺梨种质资源的遗传多样性和群体遗传结构。【拟解决的关键问题】从批量开发并合成的102对引物中,筛选出多态性最好的10对引物用于研究;基于EST-SSR标记技术,分析12个居群共255份贵州野生刺梨的遗传多样性及其居群遗传结构,以期为刺梨种质资源的科学保护和可持续利用提供理论依据。
1 材料与方法
试验于 2014—2016年在贵州省果树工程技术研究中心进行。
1.1 试验材料
选择刺梨资源分布最为丰富的 12个县作为居群代表采样点(表1、图1),根据现有分布规模决定居群样本数量,每个居群取样 12—38个不等,共 255株,居群内单个样本间距离至少50 m以上,每个采样株均进行GPS定位,并详细记录各居群经度、纬度、海拔等地理条件(表1)。每株采取幼嫩叶片10—20 g,将采样叶片就地迅速保存于自封袋,置于冰盒,带回实验室用液氮处理后存于-70℃超低温冰箱,用于DNA提取。

表1 贵州12个野生刺梨居群采样点概况Table 1 Description of the 12 sampling sites of wild R. roxburghii in Guizhou province
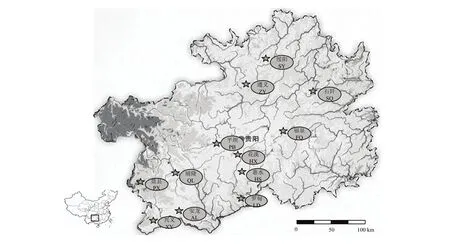
图1 贵州省12个野生刺梨居群分布情况Fig. 1 Description of the 12 sampling sites distribution of wild R. roxburghii in Guizhou province
1.2 DNA提取和SSR-PCR
1.2.1 DNA的提取 试验材料基因组DNA提取参照POREBSKI等[20]的CTAB法。
1.2.2 引物筛选 从本课题组前期已完成的批量开发的EST-SSR引物中,随机选取102对交由上海英骏生物技术公司合成。选取栽培种‘贵农5号’(Rosa roxburghii Tratt cv. GuiNong 5)、2份野生刺梨资源进行初步引物筛选。选取‘贵农5号’、15份不同来源的野生刺梨资源对筛选出的有扩增条带的引物进行多态性检测,最终确定出10对多态性较好的引物用于本研究(表2)。
1.2.3 PCR扩增 PCR反应体系为20 µL,其中包括2×Mix 10 µL;10 µmol·L-1的Primer-F、Primer-R各0.5 µL;ddH2O 8 µL;50 ng·µL-1的DNA模板1 µL。扩增程序为94℃下预变性3 min;然后进行35个循环,每个循环包括94℃变性40 s,55℃退火40 s(退火温度因不同引物而异),72℃延伸1 min;最后72℃延伸10 min。1.2.4 电泳检测 扩增产物利用8%非变性聚丙烯酰胺凝胶,于DYCZ-30C型垂直电泳槽中电泳分离,150 V电压下电泳90 min。电泳后参照BASSAM等[21]的方法进行银染显色,并在BIO-RAD凝胶成像系统中拍照记录、分析。
1.3 数据分析处理
根据分子量大小对扩增结果读带,以二倍体形式记录,从大到小依次记为 A、B、C…[22]。应用POPGENE 1.31软件[23]对各居群在多基因座位上的Hardy-Weinberg平衡进行检测,计算观测等位基因数(Na)、有效等位基因数(Ne)、多态性位点比率(P)、Nei’s基因多样性指数(Nei’s)、Shannon信息指数(I),观测杂合度(Ho)和期望杂合度(He)、基因流(Nem)、居群内近交系数(Fit)、总近交系数(Fis)、居群间分化系数(Fst),Nei’s标准遗传距离(GD)和遗传一致度(GI)。根据Nei’s标准遗传距离,利用NTSYS-pc2.10e 软件中的UPGMA方法对各居群进行聚类分析,并进行Nei’s遗传距离与地理距离的Mantel 相关性检验。
2 结果
2.1 EST-SSR标记的多态性
以‘贵农5号’和2份野生刺梨材料的基因组DNA为模板,对随机选取并合成的102对引物进行PCR扩增、筛选。结果表明,其中的71对引物产生理想的PCR产物,有效扩增率为69.61%,通过筛选确定为有效的EST-SSR引物。进一步选取‘贵农5号’和15份野生刺梨材料对71对有效引物进行扩增、多态性评价。结果31对引物呈现出多态性,占有效引物的43.66%。从31对具有多态性的引物中选取10对效果良好、结果稳定的引物(表2),对255份刺梨材料进行扩增,进而分析刺梨种质资源的遗传多样性。10对引物在255份样品上均能扩增出清晰的条带,共扩增出33个条带,不同引物的扩增条带在2—8条之间,平均3.3条,其中有27条为多态性条带,多态率为76.67%(表2)。图2为引物24在44份材料中的扩增情况。
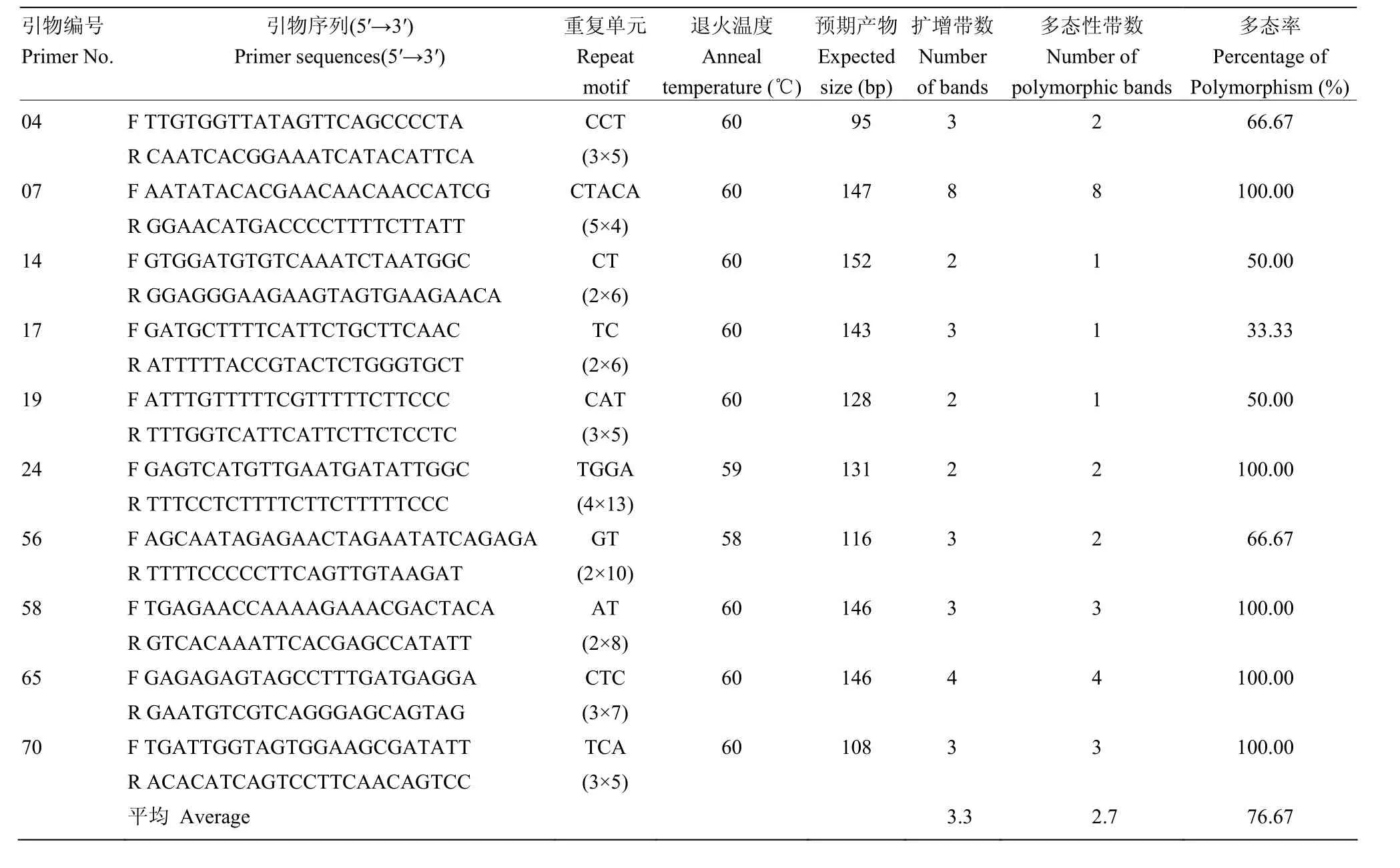
表2 用于本研究的EST-SSR引物信息Table 2 Polymorphic SSR primers used for the analyses of all 255 R. roxburghii samples
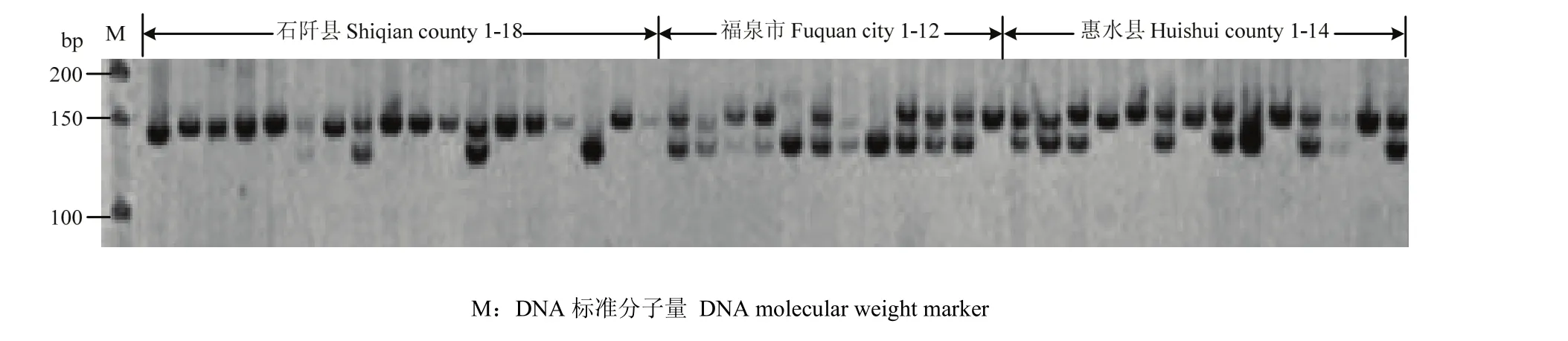
图2 引物24对刺梨基因组DNA的扩增图谱Fig. 2 SSR amplification pattern of R. roxburghii genomic DNA by primer 24
2.2 居群遗传多样性
在居群水平上,10对EST-SSR引物在12个自然居群中共扩增出33个等位基因,平均每个微卫星位点扩增 3.3个(Na=3.3)。除晴隆(QL)居群在位点17、19上,福泉(FQ)居群在位点19上只扩增出单一条带,未表现出多态性,10个居群的多态位点百分率 P为 100%(表 3),表明贵州野生刺梨具有较丰富的遗传多样性。各居群观测等位基因数 Na为 2.20—3.70;有效等位基因数 Ne为1.84—2.29;Shannon信息指数I为0.62—0.88,观测杂合度Ho为0.64—0.76;预期杂合度 He为0.44—0.54,且Ho极显著高于He(P=0.0001);惠水(HS)和花溪(HX)居群拥有最高的基因多样性指数(Nei’s为0.52),晴隆(QL)居群的基因多样性指数最低,为0.40(表3)。
2.3 居群遗传结构
采用卡方检验对各居群的多基因座位进行检测发现,除罗甸(LD)居群外,11个刺梨居群均在1/3以上的位点显著偏离 Hardy-Weinberg平衡(表4),在04位点甚至全部显著偏离。12个刺梨居群的基因多样性指数Nei’s均大于0.4;Shannon信息指数I除晴隆(QL)外均大于0.70,最高为0.88(表3)。以上结果表明,12个刺梨居群内具有较高的遗传多样性。
贵州 12个刺梨居群在 10个微卫星位点上的F-statistics分析结果见表5。12个刺梨居群在各位点上Fit、Fis为负值,Fst值在位点65最大,为0.0773;在位点4最小,为0.0092。Fst平均值为 0.0403,居群各位点基因流Nem值均大于2,且平均值为5.9484。
贵州刺梨居群间 GI为 0.9068—0.9926,平均值0.9586,遗传一致度非常高(表6);平均Nei’s遗传距离(GD)0.0431,花溪(HX)和平坝(PB)居群Nei’s遗传距离最近,仅为0.0074,兴义(XY)居群和晴隆(QL)居群之间的Nei’s遗传距离最大,也仅为0.0978。
2.4 居群聚类结果
基于贵州刺梨居群间 Nei’s遗传距离[24],采用UPGMA 法进行聚类分析,进一步直观分析居群间遗传关系,如聚类图(图3)所示,在Nei’s遗传距离0.06处可将12个贵州刺梨居群分为2个组。石阡(SQ)和兴义(XY)居群聚为第一组,其他居群聚为第二组,其中地理位置相近的花溪(HX)和平坝(PB)居群首先聚在一起,显示了最近的亲缘关系。对居群间的Nei’s遗传距离和地理距离之间的Mantel(r=0.2498,P=0.9512)检测也表明,在采集地范围,刺梨的遗传变异分布和地理位置具有显著的相关性,尤其当居群间地理距离小于500 km时,其相关关系如图4所示。
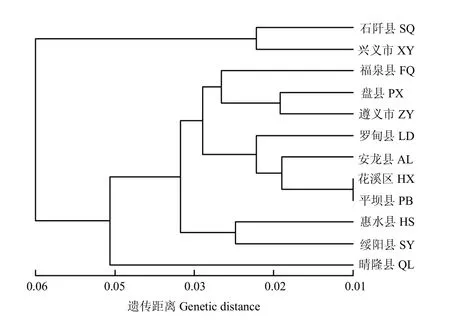
图3 基于 Nei's 遗传距离的居群UPGMA 聚类图Fig. 3 UPGMA dendrogram based on Nei’s genetic distance
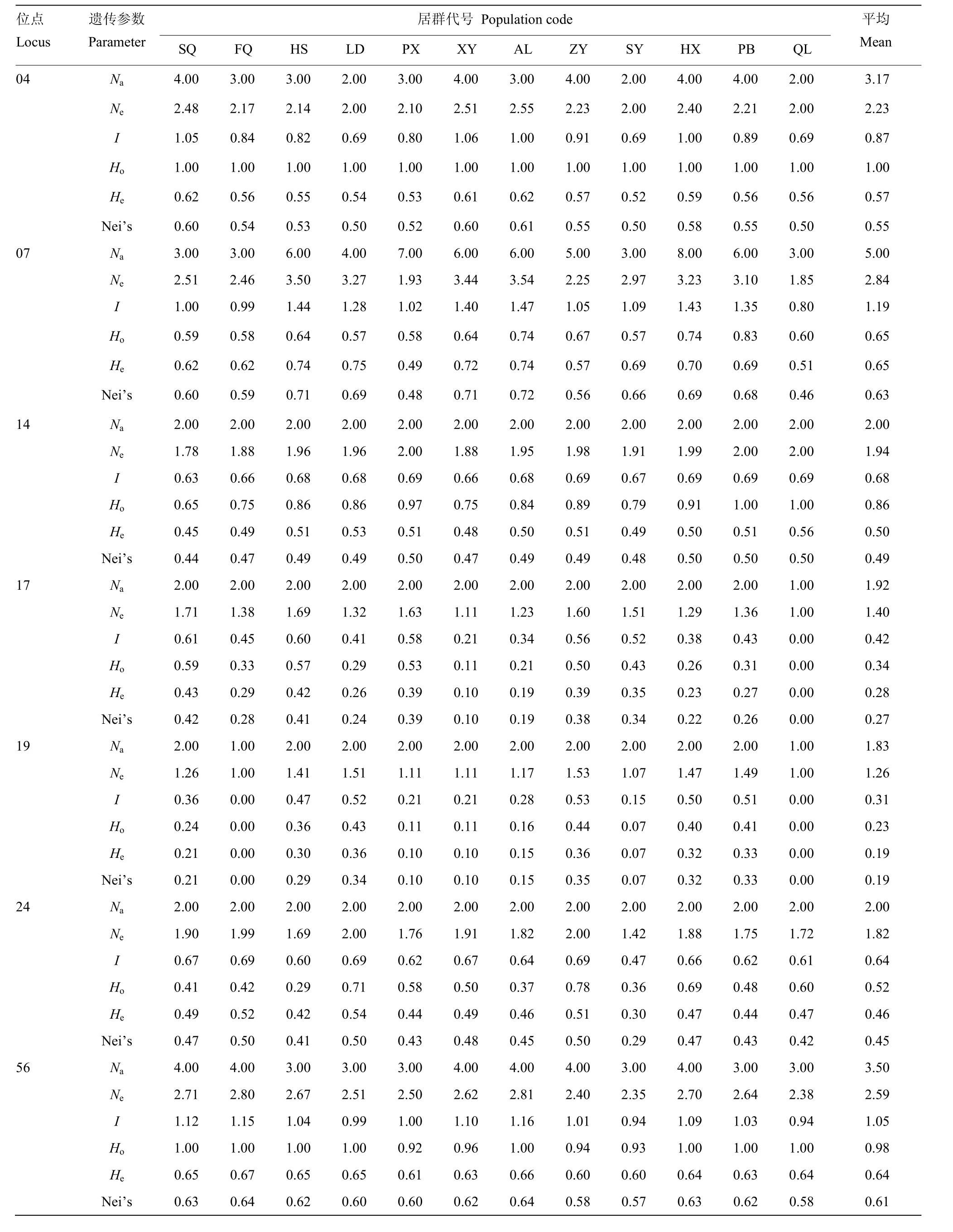
表3 贵州12个刺梨居群在10个微卫星位点的多样性指数Table 3 The genetic diversity index of 12 populations of R. roxburghii at 10 microsatellite loci

续表3 Continued table 3
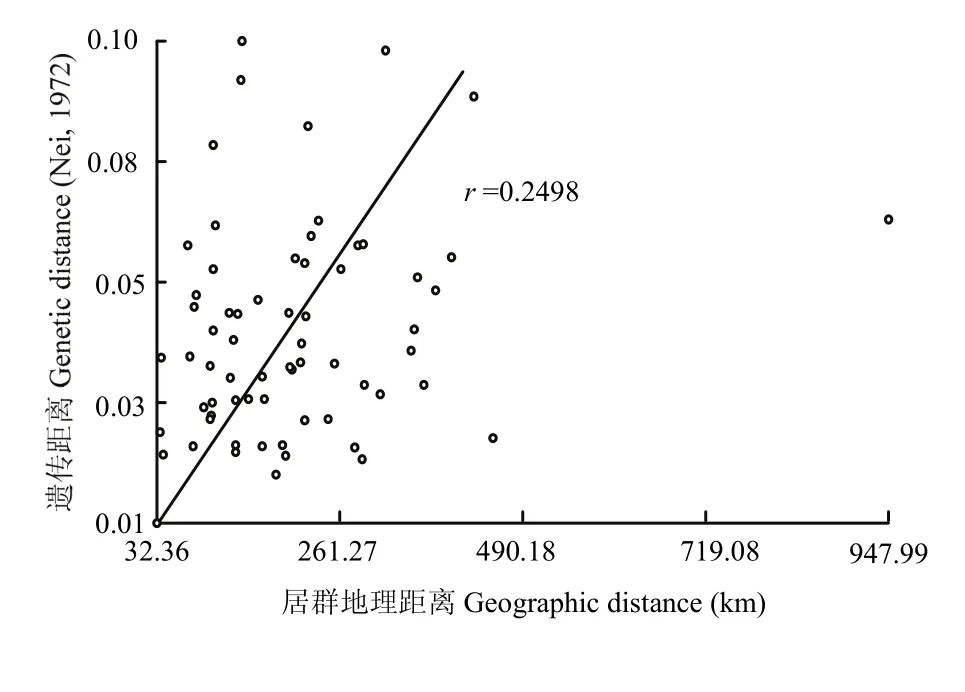
图4 12个刺梨居群间Nei’s遗传距离与地理距离的相关关系Fig. 4 The correlation between Nei’s genetic distance and geographic distance for 12 populations of R. roxburghii
3 讨论
3.1 刺梨遗传多样性
本研究利用10对EST-SSR引物,对采自贵州省12个自然居群的255份野生刺梨种质资源进行遗传多样性分析,10个微卫星位点在各居群中均表现出不同程度的多态性,除福泉(FQ)和晴隆(QL)居群外,各居群的多态位点百分率 P 均为 100%,表明这 10个微卫星位点具有高度的多态性,可作为有效的遗传标记用于刺梨居群遗传多样性和遗传结构分析。
观测等位基因数(Na)是衡量SSR位点多态性和居群变异程度高低的重要指标。10对EST-SSR引物在12个居群(255份野生刺梨种质资源)中共扩增出33个等位基因,平均每个微卫星位点扩增 3.3个(Na=3.3),与MENG等[7]采用7对SSR标记对濒危物种大花香水月季 27个居群的研究结果接近(Na=3.9),低于孙萍等[25]利用 10对SSR引物对清凉峰地区36份三叶海棠的扩增结果(Na=7.1),低于LIU等[5]利用14对SSR引物对中国浙江省77份豆梨资源的扩增结果(Na=9.5),也低于陈娇等[6]采用10对SSR引物对5个中国樱桃野生居群共133株的研究结果(Na=7.8),这可能是由于EST-SSR来源于表达的基因组区域,所以多态性比基因组SSR标记稍低[26]。
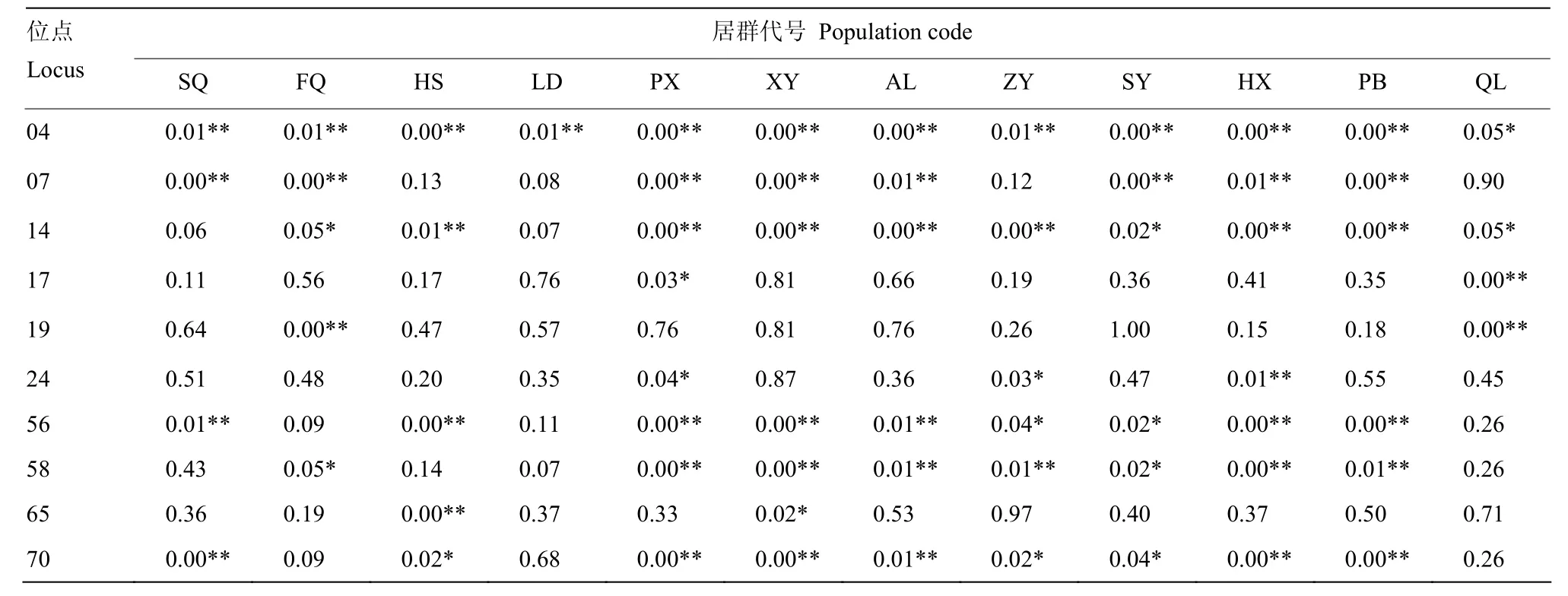
表4 Hardy-Weinberg平衡检测的P 值Table 4 The P values for Hardy-Weinberg Equilibrium test

表5 各位点固定指数及基因流Table 5 The fixed index and gene flow of 10 microsatellite loci
平均有效等位基因数(Ne)、平均Shannon信息指数(I)、平均期望杂合度(He)平均基因多样性指数(Nei’s)等是评价遗传多样性的重要指标。本试验研究结果(Ne=2.19,I=0.87,Ho=0.70,He=0.51,Nei’s=0.51),低于同属于蔷薇科的中国樱桃野生居群[6](Nei’s=0.70,I=1.53),三叶海棠[25](He=0.70,I=1.46,Ne=3.95);却高于野杏[27](Nei’s=0.29,I=0.46)和新疆野苹果[3](He=0.26,I=0.41,Ne=1.43),表明相较于蔷薇科其他植物而言,贵州野生刺梨群体遗传多样性处于适中到较高水平。这可能与刺梨的生活习性(如繁殖方式等)有关,刺梨可自花授粉结实,也可异花授粉结实,因此其遗传多样性倾向于界于杂交物种和自交物种之间。
3.2 刺梨遗传分化
贵州野生刺梨居群间分化系数 Fst平均值为0.0403,低于浙江野生豆梨[5](Fst=0.136),云南野生大花香水月季[7](Fst=0.240)及云南野生川梨[28](Fst=0.404),表明刺梨居群间属低度分化[29],居群间的遗传变异只占总变异的较少部分(4.03%);居群间基因流Nem=5.9484,表明居群间基因交流频繁,从而有效抑制了由遗传漂变而引起的遗传分化[30]。同时,频繁的基因交流必然导致居群间遗传的均质化,使各居群间表现出较高的遗传一致度(GI>0.9)及较小的遗传距离(GD<0.0978)。本试验所收集材料多集中在道路两旁,人类采集活动频繁,可能是促进刺梨居群间基因交流的重要原因。此外,也有相当部分试验材料采自人迹罕至的山坡、河沟边,因此流水和采食刺梨果实的鸟兽,也可能是刺梨远距离传播基因交流的主要方式。
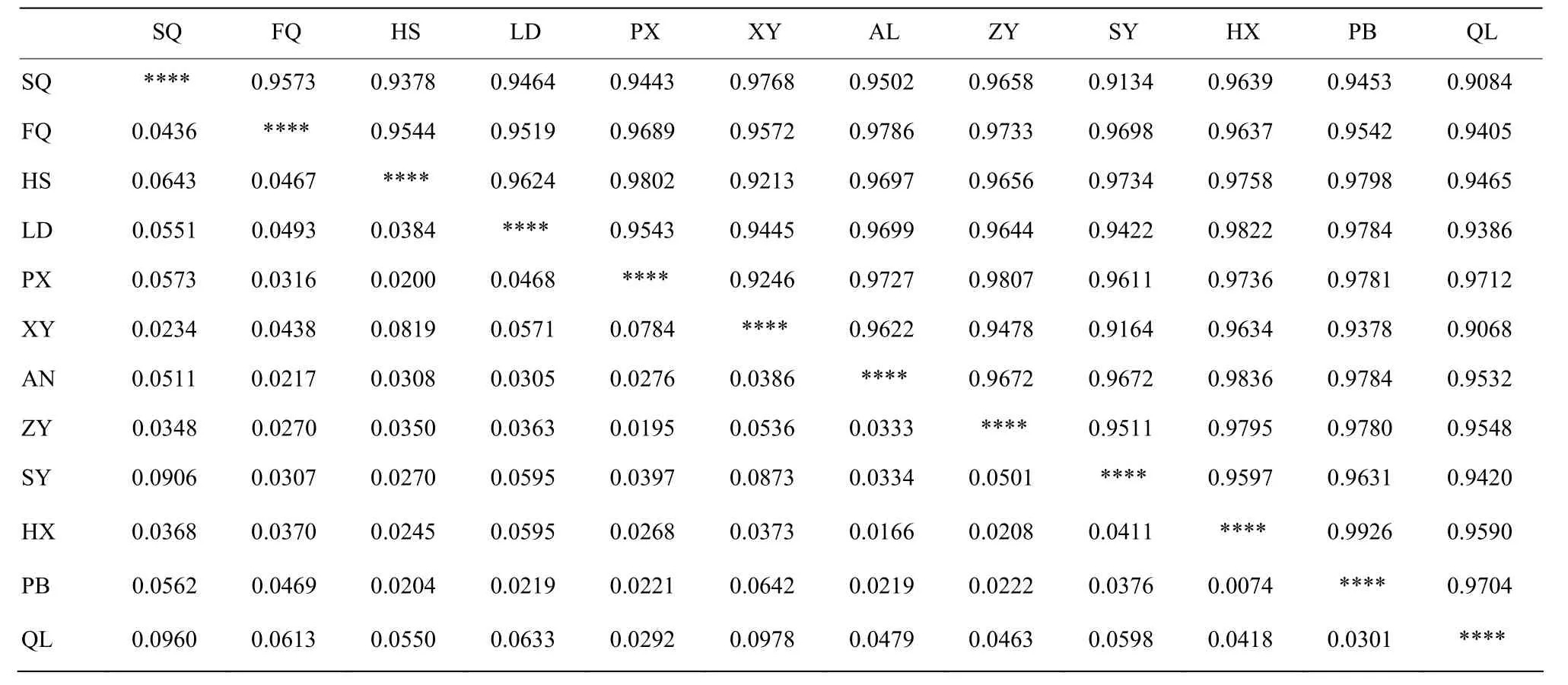
表6 居群间的Nei’s遗传距离(GD)和遗传一致度(GI)Table 6 Nei’s genetic distance and genetic identity between populations
贵州野生刺梨 12个居群在多个位点上偏离Hardy-Weinberg平衡,且观测杂合度高于期望杂合度,表明贵州野生刺梨居群内不仅遗传多样性高,还存在杂合度过剩的现象。推测可能主要存在两方面的原因:一方面,由于刺梨遗传背景较为狭窄,起源亲本数量有限,奠基者效应(Founder effect)会导致连锁不平衡现象,继而造成杂合子过剩;另一方面,可能是虫媒和风媒造成的高异花授粉率,导致刺梨野生自然居群内有性繁殖的自交率低(Fis、Fit<0),而频繁的基因交流(Nem=5.9484)使种内杂交产生的杂合子可通过无性繁殖方式进行有效固定,从而导致杂合度过剩。对居群间的Nei’s遗传距离和地理距离之间的Mantel检测表明,刺梨的遗传变异分布和地理位置具有显著的相关性,暗示地理距离可能是影响刺梨遗传分化的主要因素。
3.3 刺梨保护策略
由于土地开垦以及城市化进程等导致的土地侵占、环境破坏以及生境破碎化等,加上农业耕作时的乱砍滥伐,使得野生刺梨分布面积和群体数量急剧减少。以前在田间地头、房前屋后随处可见的刺梨资源,现在必须要深入到荒山坡地才能觅其踪影。可以预见,随着城市化发展日新月异和生态环境的日趋恶化,刺梨种质资源大量丢失的现象将日趋严重。因此,刺梨资源的收集、评价、保护等方面的工作迫在眉睫。本试验结果表明,贵州野生刺梨 12个居群中以花溪(HX)、惠水(HS)、平坝(PB)、安龙(AL)、遵义(ZY)等5个居群拥有高于平均值的观测等位基因数(Na)、有效等位基因数(Ne)、Nei’s基因多样性指数(Nei’s)、Shannon信息指数(I),观测杂合度(Ho)和期望杂合度(He)(表 3),遗传多样性较高,应当采取就地保存的策略进行种质保存,其中平坝(PB)和花溪(HX)居群遗传距离最小,且地理距离也较近,可优先选择遗传多样性更高的花溪(HX)居群进行保护;而对于人为破坏最为严重的石阡(SQ)、罗甸(LD)、盘县(PX)等3个居群则应采取迁地保存的策略。下一步将扩大野生资源的采集范围和SSR引物的筛选范围,并选择更多的分子标记技术,更加系统地开展不同区域间遗传多样性研究,为刺梨和其他蔷薇科植物的资源分类、分子标记辅助育种、遗传图谱构建等研究提供更多专一有效的分子标记。
4 结论
基于EST-SSR标记对贵州野生刺梨12个居群255份种质资源的遗传多样性分析表明,10对 EST-SSR引物具有高度的多态性,可作为有效的遗传标记用于刺梨遗传多样性和遗传结构评价。贵州省野生刺梨自然居群内的遗传多样性较高,居群内存在杂合度过剩的现象,且绝大多数的遗传变异发生在居群内(居群间分化系数Fst平均值0.0403),居群间具有基因交流频繁(基因流Nem平均值为5.9484)、遗传一致度高(GI为0.9068—0.9926)、Nei’s遗传距离小(最大遗传距离为0.0978)等特点。基于以上结果,应优先考虑对遗传多样性较高的花溪(HX)、惠水(HS)、安龙(AL)、遵义(ZY)等4个居群实施就地保存策略。
[1] 樊卫国, 夏广礼, 罗应春, 陈夏尔, 何刚. 贵州省刺梨资源开发利用及对策. 西南农业学报, 1997, 21(3): 109-115.
FAN W G, XIA G L, LUO Y C, CHEN X E, HE G. Utilization of Rosa roxburghii resource and its developing strategy in GuiZhou province. Southwest China Journal of Agricultural Sciences, 1997, 21(3): 109-115. (in Chinese)
[2] 敖芹, 谷晓平, 孟维亮. 贵州刺梨研究进展. 耕作与栽培, 2010(6): 1-7.
AO Q, GU X P, MENG W L. Research progress of Rosa roxburghii in Guizhou province. Cultivation and Farming, 2010(6): 1-7. (in Chinese)
[3] ZHANG C, CHEN X, HE T, LIU X, FENG T, YUAN Z. Genetic structure of Malus sieversii population from Xinjiang, China, revealed by SSR markers. Journal of Genetics and Genomics, 2007, 34(10): 947-955.
[4] ZONG Y, SUN P, LIU J, YUE X Y, LI K M, TENG Y W. Genetic diversity and population structure of seedling populations of Pyrus pashia. Plant Molecular Biology Reporter, 2014, 32: 644-651.
[5] LIU J, ZHENG X Y, DANIEL POTTER, HU C Y, TENG Y W. Genetic diversity and population structure of Pyrus calleryana (Rosaceae) in Zhejiang province, China. Biochemical Systematics and Ecology, 2012, 45: 69-78.
[6] 陈娇, 王小蓉, 汤浩茹, 陈涛, 黄晓姣, 梁勤彪. 基于 SSR标记的四川野生中国樱桃遗传多样性和居群遗传结构分析. 园艺学报, 2013, 40(2): 333-340.
CHEN J, WANG X R, TANG H R, CHEN T, HUANG X J, LIANG Q B. Assessment of genetic diversity and populations genetic structure in wild Chinese cherry from Sichuan province using SSR markers. Acta Horticulturae Sinica, 2013, 40(2): 333-340. (in Chinese)
[7] MENG J, HE S L, LI D Z, YI T S. Nuclear genetic variation of Rosa odorata var. gigantea (Rosaceae): Population structure and conservation implications. Tree Genetics & Genomes, 2016, 12(4): 1-14.
[8] 文晓鹏, 庞晓明, 邓秀新. 刺梨及部分近缘种形态学性状和 RAPD标记分析. 园艺学报, 2003, 30(2): 204-206.
WEN X P, PANG X M, DENG X X. A comparative study of RAPD and morphological approaches to characterize relationships of Rosa roxburghii Tratt. and its relatives. Acta Horticulturae Sinica, 2003, 30(2): 204-206. (in Chinese)
[9] WEN X P, PANG X M, DENG X X. Characterization of genetic relationships of Rosa roxburghii Tratt and its relatives using morphological traits, RAPD and AFLP markers. The Journal of Horticultural Science and Biotechnology, 2004, 79: 189-196.
[10] 唐开学, 邱显钦, 张颢, 李树发, 王其刚, 蹇洪英, 鄢波, 黄兴奇.云南蔷薇属部分种质资源的SSR遗传多样性研究. 园艺学报, 2008, 35(8): 1227-1232.
TANG K X, QIU X Q, ZHANG H, LI S F, WANG Q G, JIAN H Y, YAN B, HUANG X Q. Study on genetic diversity of some rosa germplasm in Yunnan based on SSR markers. Acta Horticulturae Sinica, 2008, 35(8): 1227-1232. (in Chinese)
[11] 邓亨宁, 高信芬, 李先源, 周洪英. 无籽刺梨杂交起源:来自分子数据的证据. 植物资源与环境学报, 2015, 24(4): 10-17.
DENG H N, GAO X F, LI X Y, ZHOU H Y. Molecular evidence for hybridization origin of Rosa×sterilis (Rosacea). Journal of Plant Resources and Environment, 2015, 24(4): 10-17. (in Chinese)
[12] FOUGÈRE-DANEZAN M, JOLY S, BRUNEAU A, GAO X, ZHANG L. Phylogeny and biogeography of wild roses with specific attention to polyploids. Annals of Botany, 2015, 115(2): 275-291.
[13] ZHU Z M, GAO X F, FOUGÈRE-DANEZAN M. Phylogeny of rosa sections Chinenses and synstylae (Rosaceae) based on chloroplast and nuclear markers. Molecular Phylogenetics & Evolution, 2015, 87: 50-64.
[14] 文晓鹏, 庞晓明, 邓秀新. 不同自然分布区刺梨遗传多样性的RAPD分析. 中国农业科学, 2003, 36(7): 823-828.
Wen X P, Pang X M, Deng X X. Genetic diversity in wild accessions of Rosa roxburghii Tratt from four provinces as revealed by RAPD analysis. Scientia Agricultura Sinica, 2003, 36(7): 823-828. (in Chinese)
[15] 文晓鹏, 邓秀新, 樊卫国. 刺梨主要基因型的RAPD鉴别. 山地农业生物学报, 2003, 22(4): 317-321.
WEN X P, DENG X X, FAN W G. Identification of genatypes of Rosa roxburghii as revealed by randomly amplified polymorphic DNA. Journal of Mountain Agriculture and Biology, 2003, 22(4): 317-321. (in Chinese)
[16] 鄢秀芹, 鲁敏, 安华明. 刺梨转录组 SSR 信息分析及其分子标记开发. 园艺学报, 2015, 42(2): 341-349.
YAN X Q, LU M, AN H M. Analysis on SSR information intranscriptome and development of molecular markers in Rosa roxburghii. Acta Horticulturae Sinica, 2015, 42(2): 341-349. (in Chinese)
[17] YAN X Q, ZHANG X, LU M, HE Y, AN H M. De Navo sequencing analysis of the Rosa roxburghii fruit transcriptome reveals putative ascorbate biosynthetic genes and EST-SSR markers. Gene, 2015, 561: 54-62.
[18] LU M, AN H M, LI L L. Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii tratt and leaf ascorbate metabolism genes. PLoS ONE, 2016, 11(2): e0147530. doi:10.1371/journal.pone.0147530.
[19] POWELL W, MACHRAY G C, PROVAN J. Polymorphism revealed by simple sequence repeats. Trends in Plant Science, 1996, 1(7): 215-222.
[20] POREBSKI S, BAILEY G, BAUM R B. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polypheNal components. Plant Molecular Biology Reporter, 1997, 15(1): 8-15.
[21] BASSAM B J, CAETANA-ANALLES G, GRESSHOFF P M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Analytical Biochemistry, 1991, 196: 80-83.
[22] 万宣伍, 刘映红, 张彬, 周浩东. 基于微卫星分子标记的重庆地区桔小实蝇遗传分化研究. 中国农业科学, 2010, 43(13): 2688-2696.
WAN X W, LIU Y H, ZHANG B, ZHOU H D. Genetic differentiation among poupulations of the oriental fruit fly Bactrocera dorsalis (Hendel) in Chongqing based on microsatellite markers. Scientia Agricultura Sinica, 2010, 43(13): 2688-2696. (in Chinese)
[23] YEH F C, YANG R C, BOYLE T. Popgene version 1.31 quick user guide. Canada: University of Alberta and Centre for International Forestry Research, 1999.
[24] NEI M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 1978, 89: 583-590.
[25] 孙萍, 宗宇, 刘晶, 胡春云, 滕元文. 基于SSR标记的清凉峰地区三叶海棠遗传多样性研究. 果树学报, 2013, 30(1): 8-15.
SUN P, ZONG Y, LIU J, HU C Y, TENG Y W. Study on genetic diversity of Malus sieboldii in Qingliangfeng region based on SSR markers. Journal of Fruit Science, 2013, 30(1): 8-15. (in Chinese)
[26] POWELL W, MACHRAY G C, PROVAN J. Polymorphism revealed by simple sequence repeats. Trends in Plant Science, 1996, 1(7): 215-222.
[27] HE T M, CHEN X S, XU Z, GAO J S, LIN P J, LIU W, LIANG Q, WU Y. Using SSR markers to determine the population genetic structure of wild apricot (Prunus armeniaca L.) in the Ily Valley of west China. Genetic Resources and Crop Evolution, 2007, 54(3): 56-572.
[28] LIU J, SUN P, ZHENG X Y, DANIEL POTTER, LI K M, HU C Y, TENG Y W. Genetic structure and phylogeography of Pyrus pashia L. (Rosaceae) in Yunnan province, China, revealed by chloroplast DNA analyses. Tree Genetics & Genomes, 2013, 9: 433-441.
[29] WRIGHT S. Evolution and the Genetics of Populations, Volume 4: Variability within and among Natural Populations. Chicago: University of Chicago Press, 1978: 65-134.
[30] SLATKIN M. Estimating levels of gene flow in natural populations. Genetics, 1981, 99: 323-335.
(责任编辑 赵伶俐)
Analysis of the Genetic Diversity of Wild Rosa roxburghii Populations in Guizhou Province Based on EST-SSR Marker
ZHANG HuaiShan1, YAN XiuQin1, LU Min1, WANG DaoPing2, AN HuaMing1
(1College of Agriculture of Guizhou University/Guizhou Engineering Research Center for Fruit Crops, Guiyang 550025;2The Key Laboratory of Chemistry for Natural Product of Guizhou Province and Chinese Academy of Sciences, Guiyang 550002)
【Objective】Genetic structure and genetic diversity of wild Rosa roxburghii resources were analyzed and evaluated in order to provide a scientific basis of the protection and excavation of R. roxburghii resources.【Method】Ten EST-SSR primers with good amplification and high polymorphism were used to analyze the genetic diversity and genetic structure within 12 populations including 255 R. roxburghii germplasms by POPGENE 1.31 software. According to Nei’s standard genetic distance, NTSYS-pc2.10e software was used to cluster the populations, and the Mantel correlation relationship between Nei's genetic distance and geographical distance was tested.【Result】The range of Shannon’s information index (I) was from 0.62 to 0.88, Nei’s gene diversity from 0.40 to 0.52. Total percentage of polymorphic loci (P) was 100% in these populations, except for Fuquan and Qinglong populations. The Chi-square test results showed that the 12 populations of R. roxburghii did not follow with the Hardy-Weinberg Equilibrium at most loci, and the Fitand Fisvalues were negative. Characteristics, such as Fst=0.0403, gene flow Nem=5.9484, genetic identity (0.9068 Rosa roxburghii; SSR; genetic diversity; population genetic structure 2016-08-12;接受日期:2016-12-02 国家自然科学基金(31660558)、贵州省高层次创新型人才培养计划(黔科合人才20164016)、贵州省科技计划项目(黔科合SY字20153026-1) 联系方式:张怀山,Tel:18798855583;E-mail:huaishan6013@163.com。通信作者安华明,E-mail:anhuaming@hotmail.com
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2022年2期)2022-03-29
大众科学(2021年10期)2021-12-23
大众科学(2021年10期)2021-12-23
浙江中医药大学学报(2021年6期)2021-07-12
中国粮油学报(2020年12期)2021-01-09
现代园艺(2017年21期)2018-01-03
中央民族大学学报(自然科学版)(2015年1期)2015-06-11
中国神经精神疾病杂志(2013年4期)2013-03-11
