
中华蜜蜂幼虫肠道参考转录组的de novo组装及SSR分子标记鉴定
2017-04-07 00:56徐细建郭睿骆群熊翠玲梁勤张串联郑燕珍张曌楠黄枳腱张璐李汶东陈大福
中国农业科学 2017年6期
徐细建,郭睿,骆群,熊翠玲,梁勤,张串联,郑燕珍,张曌楠,黄枳腱,张璐,李汶东,陈大福
(1福建农林大学蜂学学院,福州 350002;2江西省养蜂研究所,南昌 330201)
中华蜜蜂幼虫肠道参考转录组的de novo组装及SSR分子标记鉴定
徐细建1,郭睿1,骆群2,熊翠玲1,梁勤1,张串联2,郑燕珍1,张曌楠1,黄枳腱1,张璐1,李汶东1,陈大福1
(1福建农林大学蜂学学院,福州 350002;2江西省养蜂研究所,南昌 330201)
【目的】利用RNA seq技术对中华蜜蜂(Apis cerana cerana,简称中蜂)幼虫肠道参考转录组进行de novo组装,并进行功能及代谢通路注释,进而利用该转录组数据进行中蜂幼虫的SSR分子标记鉴定。【方法】实验室条件下饲养中蜂幼虫,将纯化的蜜蜂球囊菌(Ascosphaera apis,简称球囊菌)孢子饲喂3日龄幼虫,剖取 4、5和 6日龄幼虫肠道,液氮速冻。将健康幼虫肠道与感染球囊菌的幼虫肠道同时进行 Illumina测序。通过对raw reads的过滤得到clean reads,利用Trinity软件组装得到unigenes。通过BLASTx(E-value<10−5)比对NCBI Nr、Swiss-Prot、KOG和KEGG数据库,对unigenes进行功能和代谢通路注释。利用MISA软件对所有unigenes进行SSR搜索,并利用Primer Premier 5软件设计特异性SSR引物,通过常规PCR对来源于北京、辽宁兴城和四川成都的中蜂幼虫肠道样本进行SSR位点鉴定。【结果】中蜂幼虫肠道的RNA seq共得到35 670 000条reads,de novo组装得到43 557个unigenes,平均长度为898 nt。共有18 225个unigenes可被注释到上述公共蛋白数据库,单独注释到NCBI Nr、Swiss-Prot、KOG和KEGG数据库的unigenes数分别为3 899、443、37和10个。KOG注释结果显示,11 442条unigenes分布于25个基因家族,其中注释到RNA加工和修饰家族的基因数最多,达1 249个。9 679个unigenes的GO分类结果显示,在生物学进程分类中,注释到细胞进程的基因最多,达4 201个,在分子功能和细胞组分类中,注释到结合与细胞的基因数最多,分别为4 935和2 900个。4 517个unigenes可注释到KEGG数据库中的216个代谢通路,注释到核糖体的基因数最多,达385个。利用MISA软件,可在7 763个unigenes搜索到13 448个SSR位点,随机选取20对SSR引物对国内3个不同来源的中蜂幼虫肠道样本的SSR位点进行扩增,有6对引物可鉴定出SSR分子标记。【结论】成功组装并注释了中蜂幼虫肠道参考转录组,可为中蜂及其幼虫的分子生物学及组学研究提供重要的参考信息,也可用于补充、丰富和检验东方蜜蜂的参考基因组,基于此转录组数据开发出6个中蜂的SSR分子标记,可应用于中蜂的基因图谱构建、基因多样性分析、基因定位等研究,也说明利用转录组数据开发非模式生物SSRs的方法可行。
RNA seq;参考转录组;中华蜜蜂;unigene;SSR
0 引言
【研究意义】蜜蜂是重要的授粉昆虫和社会学模式昆虫,因其对研究神经生物学、发育、社会行为和表观基因组学的重要性而广受关注[1-4],西方蜜蜂(Apis mellifera)早在2006年就已完成基因组测序[5],为研究蜜蜂行为、遗传进化和基因功能提供了重要的信息和线索。与西方蜜蜂相比,东方蜜蜂(Apis cerana)更易适应极端天气、飞行距离更长、具有更强的梳理行为和清洁行为以及群体防御能力[6-9]。中华蜜蜂(Apis cerana cerana,简称中蜂)长期进化适应本土环境,相比于西方蜜蜂具有抗螨害、耐寒、善利用零星蜜粉源等优点[8-12]。利用RNA seq技术对中蜂幼虫肠道进行深度测序,de novo组装其参考转录组并进行功能及代谢通路注释,可为中蜂幼虫的分子及组学研究提供重要参考信息,在此基础上鉴定出的SSR分子标记可为在分子水平深入研究中蜂的重要性状、复杂行为及遗传进化提供宝贵信息。【前人研究进展】近年来,以RNA seq为代表的高通量测序技术发展迅猛,广泛应用于动植物及微生物研究[13-19],在蜜蜂研究方面也取得了一系列重要进展[20-21]。中国养蜂生产中的常见蜂种为意大利蜜蜂(Apis mellifera ligustica,简称意蜂)和中蜂。PARK等[22]通过对 A. cerana雄蜂的基因组测序和对A. mellifera及A. cerana工蜂多个组织的转录组测序,获得238 Mb的基因组数据和10 651个基因信息,并对A. cerana特有基因进行了分析,但作者当时并未公布基因位置及功能注释信息。SSR分子标记开发的传统方法是通过构建基因组DNA文库,成本昂贵且费时费力,而利用高通量测序技术的新一代SSR鉴定则更为经济、高效[13-15]。梁勤等[23]利用6对微卫星DNA标记对福建省4个中蜂群体进行遗传多样性分析,评估群体内的遗传变异和群体间的遗传分化;徐新建等[24]应用10个微卫星DNA标记对海南岛11个地点和大陆2个地点中蜂分析表明,海南中蜂多样性丰富,岛屿和邻近大陆种群发生了明显的遗传分化。目前,已开发的中蜂SSR分子标记很少[25-26],制约了中蜂分子进化及种群遗传学的发展。【本研究切入点】东方蜜蜂的基因组已完成测序并公布,但缺乏专一的中蜂幼虫肠道参考转录组,严重制约中蜂幼虫的病原-宿主互作及免疫应答研究。【拟解决的关键问题】利用RNA seq数据组装并注释中蜂幼虫肠道参考转录组,并鉴定出若干SSR分子标记,解决中蜂幼虫参考转录组缺失以及SSR分子标记较少的问题。
1 材料与方法
试验于2015年12月至2016年8月在福建农林大学蜂学学院蜜蜂保护学实验室完成。
1.1 供试材料
中蜂幼虫取自福建农林大学蜂学学院教学蜂场,蜜蜂球囊菌(Ascosphaera apis)菌株保存于福建农林大学蜂学学院蜜蜂保护学实验室。
1.2 主要试剂及仪器
RNase-free水购自中国上海生工生物公司;DNaseI和Oligotex mRNA Kits Midi试剂盒购自德国Qiagen公司;Dynal M280磁珠购自Invitrogen公司;高碘酸钠购自美国Sigma公司;DNA ligase购自美国Thermo公司;RNA Reagent抽提试剂盒、Ex Taq polymerase及Superscript II reverse transcriptase均购自日本TaKaRa公司;纯化cDNA的Ampure beads为美国 Agencourt产品;cDNA 文库构建试剂盒TruSeqTMDNA Sample Prep Kit -Set A为美国Illumina公司产品。其他试剂均为国产分析纯。
恒温恒湿气候箱购自中国宁波江南仪器厂;pH计购自中国上海仪电科学股份有限公司;超纯水仪购自中国四川沃特尔水处理设备有限公司;高速冷冻离心机购自德国Eppendorf公司;倒置显微镜为中国上海光学仪器五厂产品;超净工作台为中国苏州安泰空气技术有限公司产品;PCR仪为美国Bio Rad公司产品;凝胶成像系统为中国上海培清科技有限公司产品;超低温冰箱为中科美菱低温科技股份有限公司产品。
1.3 方法
1.3.1 幼虫的人工饲养 中蜂幼虫的人工饲料参照王倩等[26]的方法配制并改良,将 D-果糖和 D-葡萄糖换为新鲜蜂蜜。预试验中对照组中蜂幼虫7日龄成活率达到70%以上。从福建农林大学蜂学学院蜂场选择经PCR检测为球囊菌阴性的健康蜂群。用灭菌的移虫针挑取2日龄幼虫,放入无菌的24孔细胞培养板(每孔对应1只幼虫,孔内加有35℃预温的幼虫饲料),将24孔板放入恒温恒湿培养箱,每隔24 h吸去旧饲料、加入新饲料。3日龄时,一组幼虫饲喂含有球囊菌孢子(1×107孢子/mL)的人工饲料,另一组幼虫饲喂以正常人工饲料。35℃,90% RH条件下饲喂幼虫至7日龄,上述两组幼虫组均设置3个生物学重复。1.3.2 测序样品准备 分别于4、5、6日龄剖取中蜂幼虫肠道组织,为尽量减少肠道 RNA的降解,将从解剖取样到样品放入液氮速冻的时间控制在 30 s以内,每剖取一组样品,液氮速冻后迅速放入-80℃超低温冰箱保存。
1.3.3 cDNA文库构建及 RNA seq 利用 RNAiso Reagent试剂盒抽提中蜂幼虫肠道组织的总RNA,然后用RNase-free DNaseI去除基因组DNA残留。RNA的质量通过琼脂糖凝胶电泳和 NanoDrop ND-2000(NanoDrop,Wilmington,DE,USA)进行检测。利用Oligotex mRNA Kits Midi试剂盒说明书,纯化各样品总RNA中的mRNA。以10 μg mRNA作为模板,GsuI-oligo dT作为反转录引物,用1 000 U Superscript II reverse transcriptase在42℃下孵育1 h合成第1链cDNA;随后利用高碘酸钠氧化mRNA的5′端帽子结构,并连接生物素;通过Dynal M280磁珠筛选连接了生物素的mRNA/cDNA,并通过碱裂解释放第1链cDNA;然后通过DNA ligase在第1链cDNA 的5′末端加上接头,利用Ex Taq polymerase合成第2链cDNA。最后,通过GsuI酶切去除polyA和5′端接头。利用Ampure beads对上述cDNA进行纯化,cDNA文库通过TruSeqTMDNA Sample Prep Kit-Set A进行构建和TruSeq PE Cluster Kit进行扩增。委托广州基迪奥生物技术有限公司对上述12个肠道样品进行深度测序,测序平台为Illumina Hiseq 2500,各肠道样品的3个生物学重复均同时进行测序。
1.3.4 中蜂幼虫肠道参考转录组的de novo组装 首先,利用Perl脚本去除含有adaptor、未知核苷酸比例>5%和低质量 reads,获得 clean reads。利用软件Trinity[27]进行中蜂转录组的 de novo组装(缺省值Kmer=25)。长度短于200 bp的contigs和unigenes将被舍弃。过滤和组装以后得到高质量的unigenes。
1.3.5 Unigenes注释 利用 BLASTx(E-value<10−5)将测序序列比对NCBI nr数据库(http://www.ncbi. nlm.nih.gov)、Swiss-Prot 数据库(http://www.expasy.ch/ sprot)、KOG(Clusters of orthologous groups for eukaryotic complete genomes)数据库(ftp://ftp.ncbi.nih. gov/pub/COG/KOG/kyva)和KEGG代谢通路(pathway)数据库(http://www.genome.jp/kegg/)。利用BLASTX将组装出来的unigenes序列与Nr数据库进行比对后,取每个unigenes在Nr库中比对结果最好(E值最低)的那一条序列为对应同源序列(如有并列,取第一条)确定同源序列所属物种,统计比对到各个物种的同源序列数量。基于Nr database注释结果,利用Blast2GO进行unigenes的GO注释,利用WEGO软件对每一个转录本进行GO分类。
1.3.6 SSR分子标记开发 利用软件 MISA(http:// pgrc.ipk-gatersleben.de/misa/)搜索unigenes的微卫星标记,按照以下标准从unigenes中查找SSR位点:二核苷酸重复≥6次,三核苷酸重复≥5次,四核苷酸重复≥5次,五核苷酸重复≥5次和六核苷酸重复≥5次。根据 MISA的输出结果,利用 Primer Premier 5(PREMIER Biosofe Int.,Palo Alto,CA)对每一个含有16 bp碱基重复的SSR设计引物。
选取北京(B)、辽宁兴城(L)、四川成都(S)3个不同来源的中蜂幼虫肠道样本作为模板,随机选取20对SSR引物进行PCR扩增,PCR程序:94℃预变性5 min;94℃变性50 s,55℃退火30 s,72℃延伸30 s,共33个循环,72℃再延伸10 min。PCR产物经1%琼脂糖凝胶电泳检测。
2 结果
2.1 中蜂幼虫肠道的RNA seq及参考转录组de novo组装
对上述12个肠道样品进行Illumina测序,平均得到30 584 420条原始读段(raw reads),去除低质量和含有接头的reads后平均获得29 726 139条有效读段(clean reads),总测序长度为3 715 767 396, Q20平均为98.31%,说明测序数据质量较好,可用于下一步分析。各样品的测序详细信息如附表1所示。
对 clean reads进行进一步序列拼接和去冗余处理,组装得到43 557条unigenes,平均长度达898 nt, N50为1 704 nt(表1)。统计结果显示,unigenes的数目随着序列长度的增加而减少,在200—299 nt长度范围内数目最多,符合生物体序列长度分布的基本规律。长度>1 000 nt的unigenes有10 454条,占总unigenes的24.00%。上述结果说明中蜂幼虫肠道的组装质量较好。转录组测序数据已上传NCBI SRA数据库,SRA号:SRA456721。
2.2 Unigenes注释
利用 BLASTx(E-value<10−5)将测序序列比对NCBI Nr、Swiss-Prot、KOG和KEGG pathway数据库,结果显示分别有17 456、12 830、11 442和9 045个unigenes能够注释到上述数据库,有功能或代谢通路注释的unigenes数目为18 225,占全部unigenes的41.84%,此外,有58.16%的unigenes无功能注释(表2)。有29个unigenes在4大数据库均有注释,而仅能注释到NCBI Nr、Swiss-Prot、KOG和KEGG pathway数据库的unigenes分别为3 899、443、37和10个。

表1 中蜂幼虫肠道参考转录组组装结果统计Table 1 Summary of A. c. cerana larval gut’s reference transcriptome assembled in this study

表2 公共蛋白数据库注释统计表Table 2 Summary of annotation information of all unigenes in public protein databases
注释到Nr数据库中unigenes的E-value分布显示(图1),比对到物种序列的E-value均<10-5,其中E-value<10-100的有49.76%,说明比对结果可信度较高。注释基因同源序列的物种分布统计结果显示前 10位的物种依次为 Apis mellifera、Apis dorsata、Apis florea、Bombus impatiens、Bombus terrestris、Lasius niger、Megachile rotundata、 Harpegnathos saltator、Capsaspora owczarzaki ATCC 30864和Cerapachys biroi,注释到A. mellifera的基因数为5 753(31.57%),注释到A. dorsata和A. florea的基因数分别为3 695(20.27%)和2 489(13.66%)(表3)。
KOG注释结果显示,11 442个unigenes分布于25个基因家族(图2)。其中,注释基因数最多的为信号转导机制,其次为一般功能预测和翻译后修饰、蛋白翻转和分子伴侣。值得注意的是,有 170条 unigenes注释到防御机制,它们可能在中蜂幼虫抵御病原入侵过程发挥重要作用。
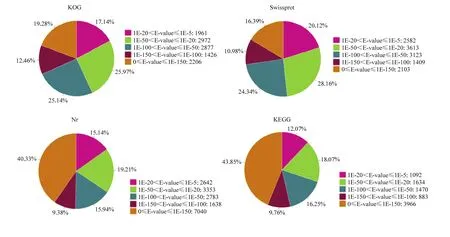
图1 E值分布Fig.1 Distribution of E-value in four databases
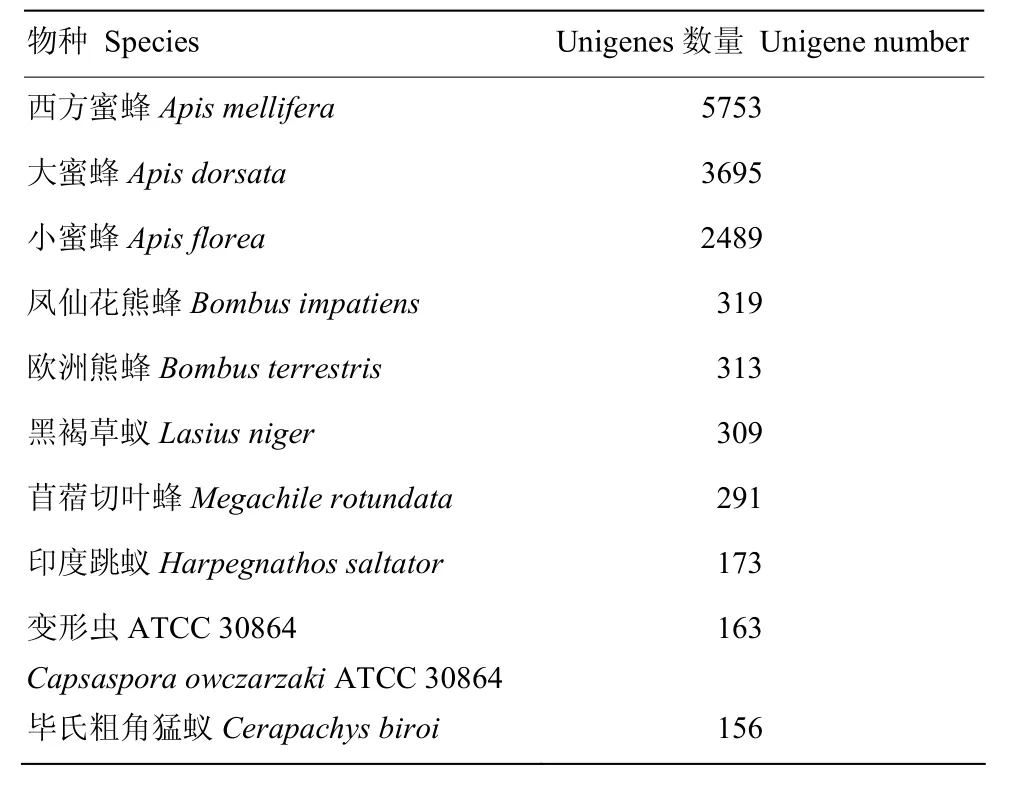
表3 Unigenes的物种分布统计表(前10位)Table 3 Unigenes distribution in different species (top 10 species)
2.3 Unigenes的Gene Ontology(GO)分类
对所有unigenes进行GO分类,共有9 679个unigenes具有GO功能注释,这些基因的功能分为生物学过程、细胞组分和分子功能3类。如图3所示,生物学进程中,注释到行为、生物黏附、生物调控、细胞杀伤、细胞成分组织或生物合成、细胞进程、生长、免疫系统进程、定位、运动、代谢进程多组织进程、多细胞组织进程、生殖、生殖进程、应激、信号、单一有机体进程的unigenes数目分别为22、92、1 655、2、519、4 156、7、16、1 132、36、4 146、52、220、25、31、819、593和3 263个;细胞组分中,注释到细胞、细胞连接、细胞零、细胞外基质、细胞外基质组分、胞外区、胞外区零件、大分子复合物、细胞膜、细胞膜零件、细胞膜内腔、细胞器、细胞器零件、突触、突触零件、病毒、病毒零件的unigenes数目分别为2 900、15、2 900、39、6、27、26、1 287、1 702、1 511、65、1 893、700、41、37、49和49;分子功能中,注释到抗氧化活性、结合、催化活性、通道调节子活性、电子转运活性、酶调节活性、脒基核苷酸交换因子活性、分子功能调节因子、分子转换器活性、核酸结合转录因子活性、蛋白结合转录因子活性、结构分子活性、转运子活性的unigenes数目分别为48、4 935、2、34、89、61、150、316、177、30、402和521。
2.4 Unigenes的KEGG代谢通路注释
对所有unigenes进行KEGG代谢通路注释,共有4 517个 unigenes注释到 KEGG数据库中,这些unigenes的通路信息如图4所示。这些unigenes分布于216个已知的代谢通路中,其中富集数量最多的10个代谢通路是核糖体、碳代谢以及内质网蛋白加工、内吞、RNA转运、嘌呤代谢、氧化磷酸化、剪接体、氨基酸生物合成和泛素介导的蛋白水解(表 4)。此外,注释到溶酶体、MAPK信号通路、Jak-STAT信号通路、昆虫激素生物合成、黑化作用、Ras信号通路、凋亡和嗅觉转化上的unigenes分别为119、27、25、16、10、7、4和4个,其中富集在免疫通路上的unigenes有可能在中蜂幼虫响应病原微生物入侵的免疫应答过程中发挥关键作用。
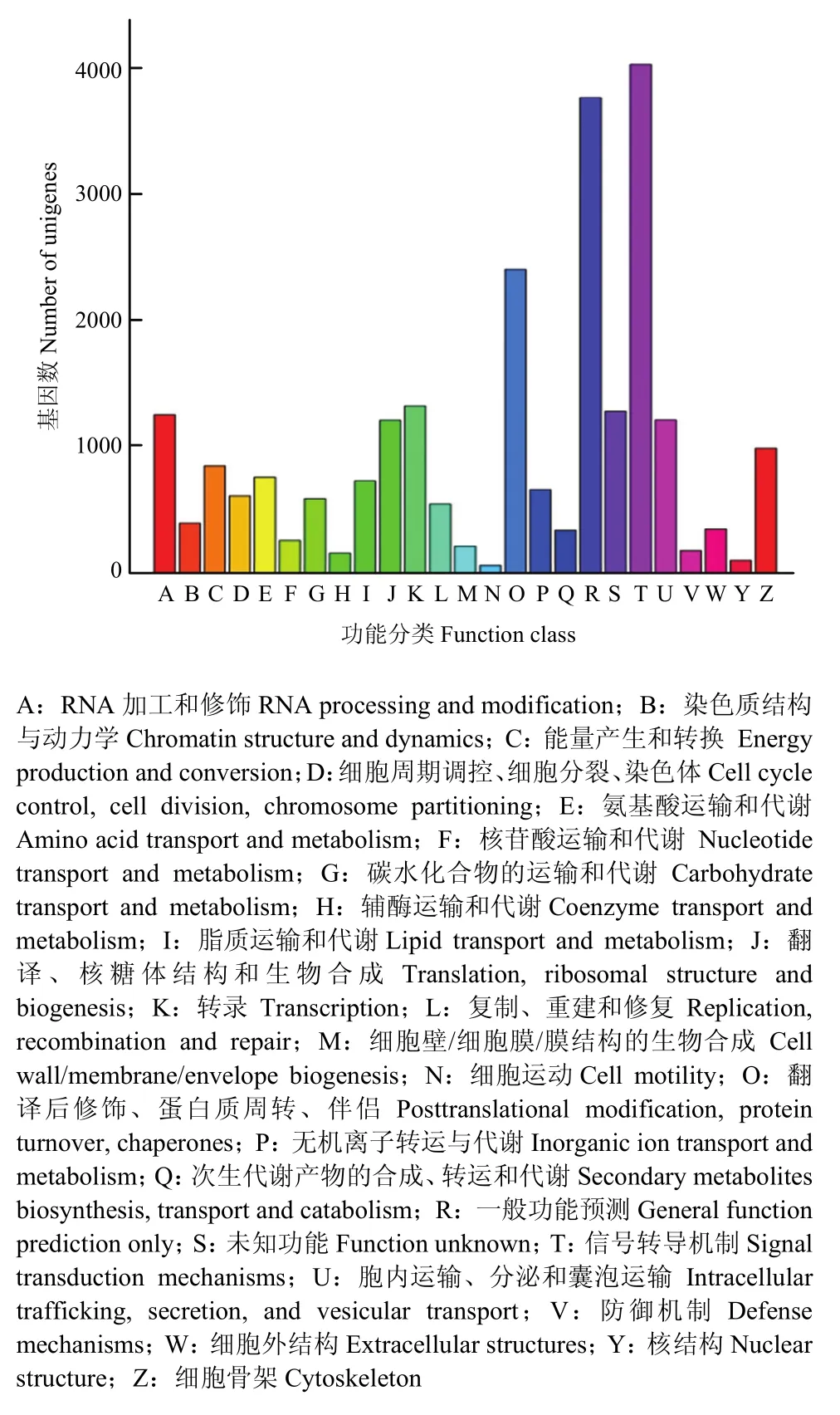
图2 Unigenes的KOG功能分类Fig.2 KOG classification of unigenes
2.5 SSR分子标记鉴定
利用MISA软件从43 557条 unigenes中共鉴定出13 448个SSR位点。其中二核苷酸重复最多,数目达到 7 804(58.03%),其次依次为三核苷酸、四核苷酸、五核苷酸和六核苷酸重复,数目分别为 3 797(28.23%)、1 307(9.72%)、339(2.52%)和201(1.49%)(表5)。通过对SSR基元进行分析,发现AT/AT出现的频率最高(30.4%),其次为AG/CT(22%),不同类型的SSR在总SSR中所占的比例如图5所示。
在上述的 13 448个 SSR位点中,利用 Primer Primer 5软件在随机挑选的20个SSRs序列两侧设计特异性引物,引物序列信息如附表2所示。提取4、5、6日龄中蜂幼虫肠道总DNA,等摩尔混合作为模板进行PCR扩增。
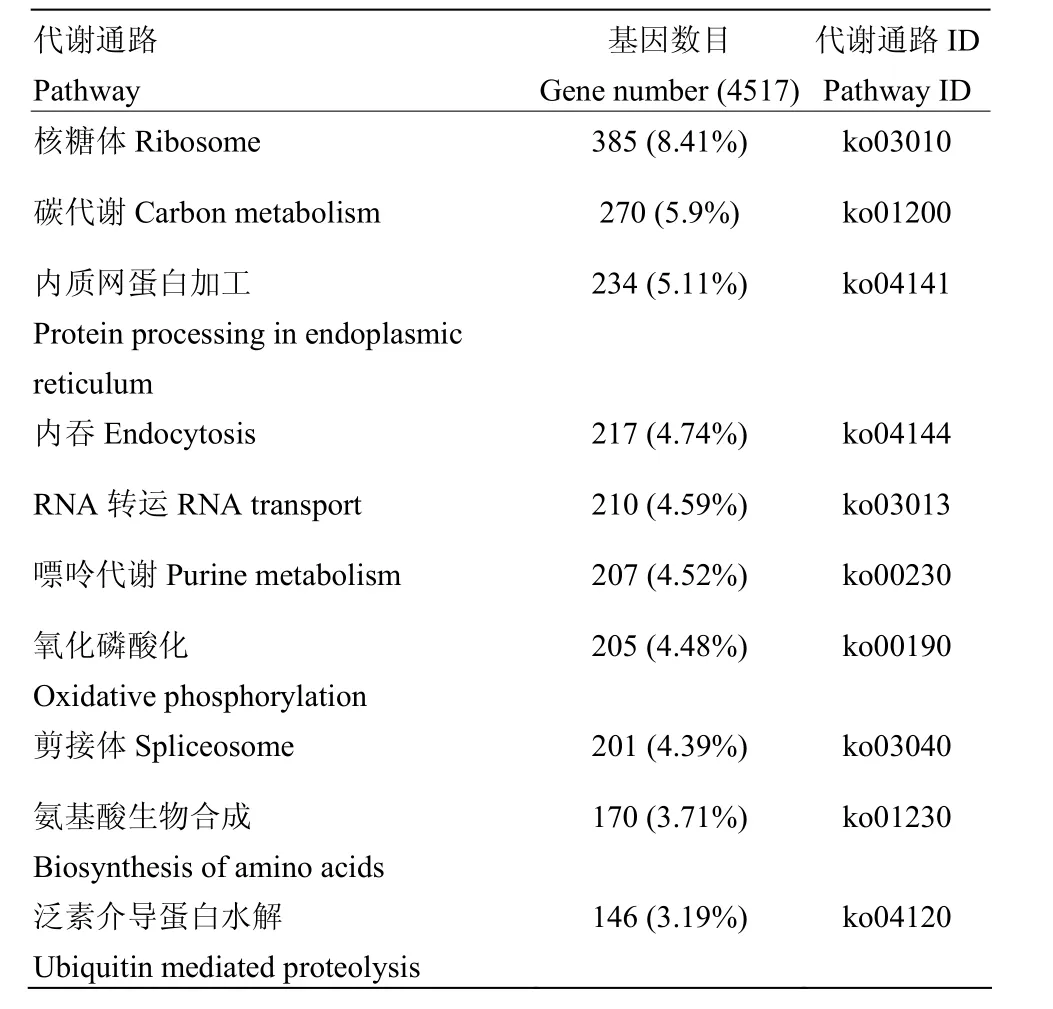
表4 注释到KEGG数据库前10位代谢通路Table 4 Top 10 pathways of unigenes annotated in KEGG pathway database
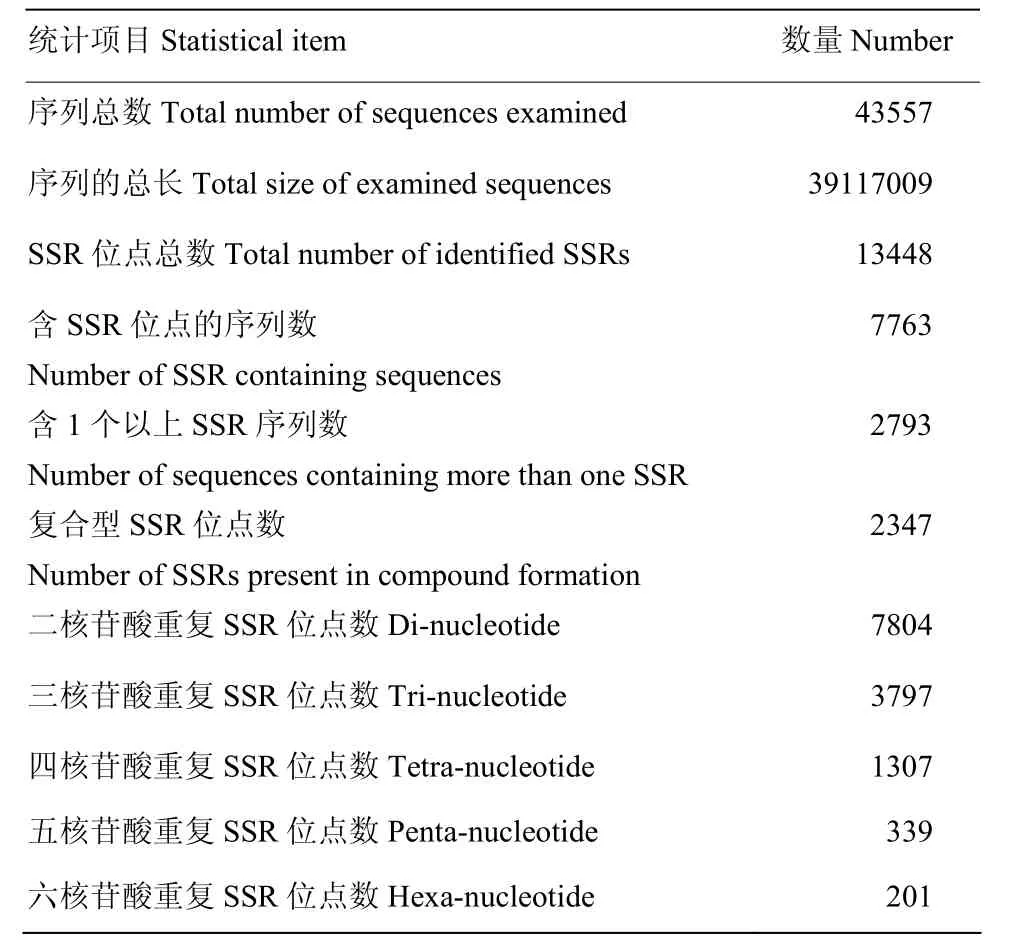
表5 中蜂幼虫肠道SSR位点统计Table 5 Characteristics of SSRs in A. c. cerana larval gut
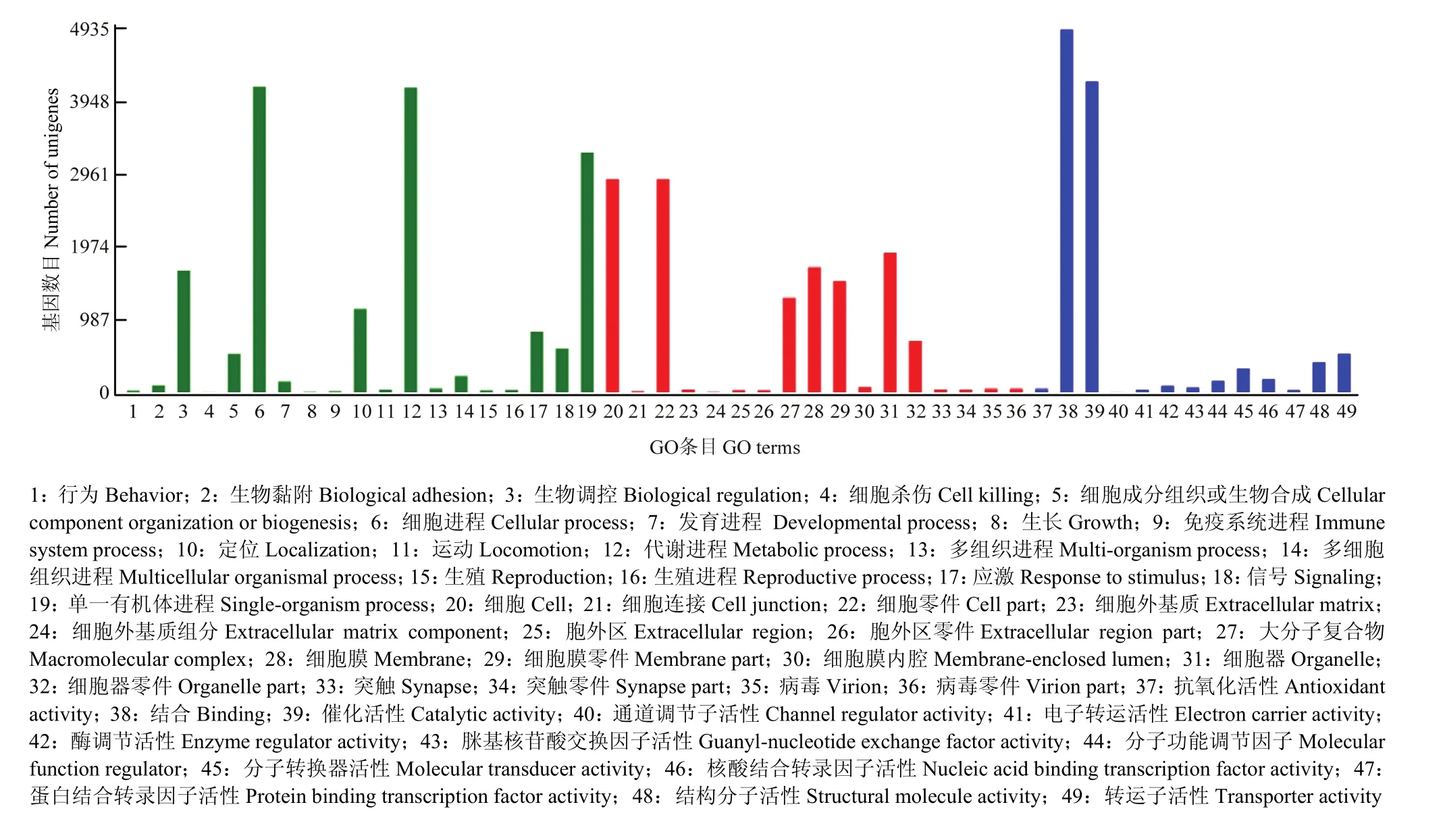
图3 Unigenes的GO分类Fig.3 GO classification of all unigenes
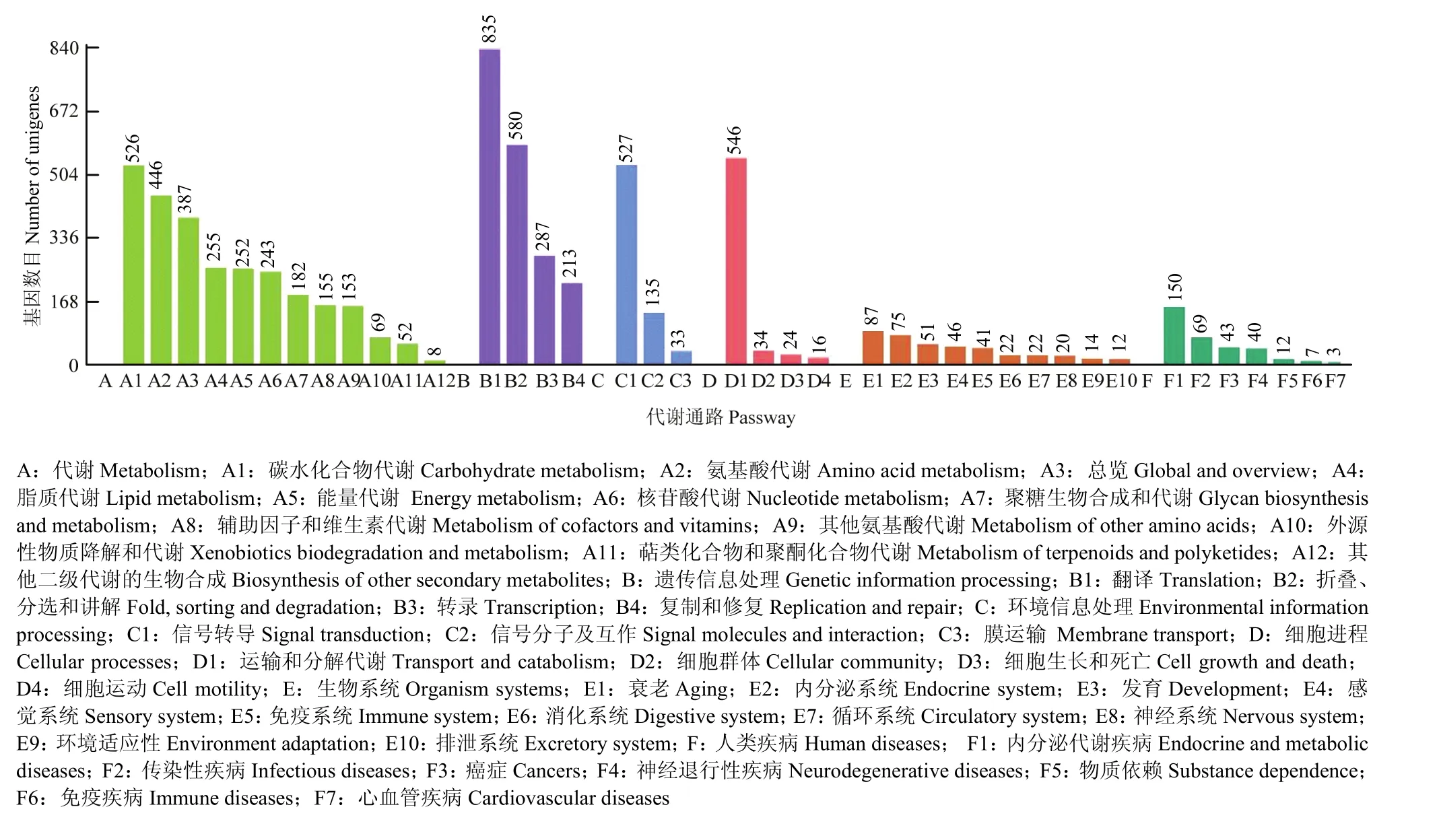
图4 Unigenes的KEGG代谢通路注释Fig.4 KEGG pathway annotation of all unigenes
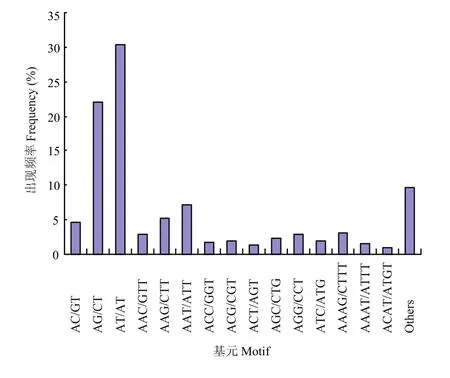
图5 不同串联重复单元类型的SSR在总SSR中所占比例Fig.5 Frequency of SSR motif in total SSRs
PCR产物经1%琼脂糖凝胶电泳检测,结果显示,有6对SSRs特异性引物(SSR9、SSR11、SSR14、 SSR16、SSR19、SSR20)对3个不同来源的中蜂幼虫肠道样品都扩增出了具有多态性的特异性条带(图6),说明这些SSR位点有望作为中蜂幼虫特有的分子标记,基于转录组数据大规模开发SSR分子标记具有良好的前景。
3 讨论

图6 国内3个来源中蜂幼虫肠道SSR位点鉴定Fig.6 SSR loci identification of A. c. cerana larval gut samples from three different regions in China
2015年,韩国的研究人员公布了东方蜜蜂雄蜂的基因组信息,但当时并没有公布基因的位置及功能注释信息[22]。WANG等[16]曾对中蜂进行过转录组测序,因测序组织包括 3日龄工蜂幼虫、1日龄工蜂蛹、1日龄成年工蜂、采集蜂以及哺育蜂,故该转录组信息较为复杂、不够专一。肠道是中蜂幼虫的主要免疫器官,在抵御病原微生物入侵过程中扮演着重要角色。本研究利用RNA seq技术对中蜂肠道进行深度测序,成功组装并注释了专一的中蜂幼虫肠道参考转录组,将有力推动中蜂及其幼虫的分子及组学研究,如中蜂幼虫响应球囊菌或东方蜜蜂微孢子虫(Nosema ceranae)侵染过程中的免疫应答及分子调控研究。
养蜂生产中,意蜂幼虫易被球囊菌感染而罹患白垩病[28],而中蜂幼虫具有较强的球囊菌抗性,但偶尔可见患病幼虫。通常认为中蜂具有较强的清理行为,表现出更强的群体防御[7],但中蜂幼虫个体水平的免疫防御却鲜有研究,其在中蜂幼虫球囊菌抗性方面所发挥的作用值得深入探讨。未来笔者课题组将在本研究组装并注释的参考转录组的基础上,对病原胁迫过程中中蜂幼虫的病原-宿主互作机制、免疫应答机制及分子调控机制进行深入系统的研究。
SSR分子标记的传统开发方法是通过构建 DNA文库进行筛选,成本高且效率低,而高通量测序技术的应用为大规模筛选 SSR分子标记带来曙光[15]。目前,已报道的中蜂SSR分子标记非常少[24-25],严重阻碍中蜂的品种鉴定及遗传进化等研究。本研究基于中蜂幼虫肠道的转录组数据预测潜在的SSR分子标记,随机选取的20对特异性SSR引物中有6对可在北京、辽宁兴城和四川成都3个不同来源的中蜂幼虫样品中扩增出具有多态性的片段,这些新开发的SSR分子标记有助于中蜂的基因图谱构建、基因多样性分析、基因定位等[29-30]研究的深入开展,说明基于转录组测序数据大规模开发SSR分子标记具有良好的应用前景。
4 结论
成功组装中蜂幼虫肠道参考转录组并对其进行了功能及代谢通路注释,可为中蜂幼虫的分子及组学研究提供重要的参考信息,也可用于补充、丰富和检验已公布的东方蜜蜂基因组,基于该转录组数据开发出的6个中蜂的SSR分子标记可应用于中蜂的基因图谱构建、基因多样性分析、基因定位等研究,同时也说明利用转录组数据开发非模式生物SSRs的方法可行。
[1] ZAYED A, ROBINSON G E. Understanding the relationship between brain gene expression and social behavior: lessons from the honey bee. Annual Review of Genetics, 2012, 46: 591-615.
[2] BEGNA D, HAN B, FENG M, FANG Y, LI J K. Differential expressions of nuclear proteomes between honeybee (Apis mellifera L.) queen and worker larvae: a deep insight into caste pathway decisions. Journal of Proteome Research, 2012, 11(2): 1317-1329.
[3] FORET S, KUCHARSKI R, PELLEGRINI M, FENG S, JACOBSEN S E, ROBINSON G E, MALESZKA R. DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(13): 4968-4973.
[4] GALIZIA G E D, GIU M. Honeybee Neurobiology and Behavior: A Tribute to Randolf Menzel. Springer, 2012: 521.
[5] WEINSTOCK G M, ROBINSON G E, GIBBS R A, WEINSTOCK G M. Insights into social insects from the genome of the honeybee Apis mellifera. Nature, 2006, 443(7114): 931-949.
[6] PENG Y S, NASR M E, LOCKE S J. Geographical races of Apis cerana Fabricius in China and their distribution. Review of recent Chinese publications and a preliminary statistical analysis. Apidologie, 1989, 20(1): 9-20.
[7] LIN Z, PAGE P, LI L, QIN Y, ZHANG Y, HU F, NEUMANN P, ZHENG H, DIETEMANN V. Go east for better honey bee health: Apis cerana is faster at hygienic behavior than A. mellifera. PLoS ONE, 2016, 11(9): e0162647.
[8] FRIES I, WEI H, SHI W, CHEN S J. Grooming behavior and damaged mites (Varroa jacobsoni) in Apis cerana cerana and Apis mellifera ligustica. Apidologie, 1996, 27(1): 3-11.
[9] 周冰峰, 朱翔杰. 论中华蜜蜂种质资源的保护//中国蜂业科技可持续发展学术研讨会暨蜂业科技与生态研讨会, 2008: 110-117.
ZHOU B F, ZHU X J. The protection of Apis cerana cerana//China Apicultural Conference on Sustainable Development & Apicultural Technology and Ecology, 2008: 110-117. (in Chinese)
[10] RATH W. Co-adaptation of Apis cerana Fabr. and Varroa jacobsoni Oud. Apidologie, 1999, 30(2/3): 97-110.
[11] PENG Y S, FANG Y, XU S, GE L. The resistance mechanism of the Asian honey bee, Apis cerana Fabr. to an ectoparasitic mite, Varroa jacobsoni Oudemans. Journal of Invertebrate Pathology, 1987, 49(1): 54-60.
[12] 龚一飞, 张其康. 蜜蜂分类与进化. 福州: 福建科学技术出版社, 2000: 21-26.
GONG Y F, ZHANG Q K. Classification and Evolution of Apis. Fuzhou: Fujian Science and Technology Press, 2000: 21-26. (in Chinese)
[13] 李红亮. 中华蜜蜂头部ESTs文库构建和主要触角特异蛋白基因克隆、定位及其表达鉴定[D]. 杭州: 浙江大学, 2007.
LI H L. Construction of cDNA libraries from head of bees, cloning, expression and subcellular localization of the antenna specific protein gene in the Chinese honeybee, Apis cerana cerana[D]. Hangzhou: Zhejiang University, 2007. (in Chinese)
[14] NOVAES E, DROST D R, FARMERIE W G, PAPPAS G J, GRATTAPAGLIA D, SEDEROFF R R, KIRST M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics, 2008, 9(1): 312.
[15] GUO R, WANG S, XUE R, CAO G, HU X, HUANG M, ZHANG Y, LU Y, ZHU L, CHEN F, LIANG Z, KUANG S, GONG C. The gene expression profile of resistant and susceptible Bombyx mori strains reveals cypovirus-associated variations in host gene transcript levels. Applied Microbiology and Biotechnology, 2015, 99(12): 5175-5187.
[16] WANG Z, GERSTEIN M, SNYDER M. RNA-seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics, 2009, 10(1): 57-63.
[17] HAAS B J, PAPANICOLAOU A, YASSOUR M, GRABHERR M, BLOOD P D, BOWDEN J, COUGER M B, ECCLES D, LI B, LIEBER M. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nature Protocols, 2013, 8(8): 1494-1512.
[18] VAN DIJK E L, AUGER H, JASZCZYSZYN Y, THERMES C. Ten years of next-generation sequencing technology. Trends in Genetics, 2014, 30(9): 418-426.
[19] TRAPNELL C, WILLIAMS B A, PERTEA G, MORTAZAVI A, KWAN G, VAN BAREN M J, SALZBERG S L, WOLD B J, PACHTER L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during celldifferentiation. Nature Biotechnology, 2010, 28(5): 511-515.
[20] WANG Z L, LIU T T, HUANG Z Y, WU X B, YAN W Y, ZENG Z J. Transcriptome analysis of the Asian honey bee Apis cerana cerana. PLoS ONE, 2012, 7(10): e47954.
[21] CORNMAN R S, BENNETT A K, MURRAY K D, EVANS J D, ELSIK C G, ARONSTEIN K. Transcriptome analysis of the honey bee fungal pathogen, Ascosphaera apis: implications for host pathogenesis. BMC Genomics, 2012, 13: 285.
[22] PARK D, JUNG J W, CHOI B S, JAYAKODI M, LEE J, LIM J, YU Y, CHOI Y S, LEE M L, PARK Y. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics, 2015, 16: 1.
[23] 梁勤, 张立卿. 利用微卫星标记分析福建4个中华蜜蜂群体的遗传多样性. 福建农林大学学报 (自然科学版), 2009, 38(4): 388-392.
LIANG Q, ZHANG L Q. Analysis of genetic diversity of four Apis cerana cerana populations in Fujian Province with microsatellite markers. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2009, 38(4): 388-392. (in Chinese)
[24] 徐新建, 周姝婧, 朱翔杰, 周冰峰. 海南岛中华蜜蜂遗传多样性的微卫星DNA分析. 昆虫学报, 2013, 56(5): 554-560.
XU X J, ZHOU S J, ZHU X J, ZHOU B F. Microsatellite DNA analysis of genetic diversity of Apis cerana cerana in Hainan Island, southern China. Acta Entomologica Sinica, 2013, 56(5): 554-560. (in Chinese)
[25] 于瀛龙, 周姝婧, 徐新建, 朱翔杰, 周冰峰. 长白山中华蜜蜂(Apis cerana cerana)遗传多样性分析. 福建农林大学学报 (自然科学版), 2013, 42(6): 643-647.
YU Y L, ZHOU S J, XU X J, ZHU X J, ZHOU B F. Analysis on genetic diversity of Apis cerana cerana in Changbai Mountains. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2013, 42(6): 643-647. (in Chinese)
[26] 王倩, 孙亮先, 肖培新, 刘锋, 康明江, 胥保华. 室内人工培育中华蜜蜂幼虫技术研究. 山东农业科学, 2009(11): 113-116.
WANG Q, SUN L X, XIAO P X, LIU F, KANG M J, XU B H. Study on technology for indoor artificial feeding of Apis cerana cerana larvae. Shandong Agricultural Sciences, 2009(11): 113-116. (in Chinese)
[27] GRABHERR M G, HAAS B J, YASSOUR M, LEVIN J Z, THOMPSON D A, AMIT I, XIAN A, FAN L, RAYCHOWDHURY R, ZENG Q. Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nature Biotechnology, 2011, 29(7): 644-652.
[28] GUPTA R K, REYBROECK W. Honeybee Pathogens and Their Management, 2014: Springer, Netherlands.
[29] SOMRIDHIVEJ B, WANG S, SHA Z, LIU H, QUILANG J, XU P, LI P, HU Z, LIU Z. Characterization, polymorphism assessment, and database construction for microsatellites from bac end sequences of channel catfish (Ictalurus punctatus): A resource for integration of linkage and physical maps. Aquaculture, 2008, 275(1/4): 76-80.
[30] GAO X, HAN J, LU Z, LI Y, HE C. Retracted: characterization of the spotted seal phoca largha transcriptome using Illumina paired-end sequencing and development of SSR markers. Comparative Biochemistry and Physiology-Part D: Genomics and Proteomics, 2012, 7(3): 277-284.
(责任编辑 岳梅)
De novo Transcriptome Assembly for Apis cerana cerana Larval Gut and Identification of SSR Molecular Markers
XU XiJian1, GUO Rui1, LUO Qun2, XIONG CuiLing1, LIANG Qin1, ZHANG ChuanLian2, ZHENG YanZhen1, ZHANG ZhaoNan1, HUANG ZhiJian1, ZHANG Lu1, LI WenDong1, CHEN DaFu1
(1College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002;2Apiculture Institute of Jiangxi Province, Nanchang 330201)
Abstract:【Objective】 The objective of this study is to de novo assemble a reference transcriptome for Apis cerana cerana larval gut, perform gene function and pathway annotation for this transcriptome, and to identify specific SSR molecular markers for A. c. cerana larvae. 【Method】 3-day-old instar A. c. cerana larvae were fed with the purified Ascosphaera apis spores, the guts of 4-, 5- or 6-day-old honeybee larvae were sampled and used as sequencing material for RNA seq. After filtration, clean reads were obtained, and unigenes were assembled using Trinity software. BLASTX tool (E-value<10−5) was used to search the unigenes against NCBI Nr, Swiss-Prot protein, KOG as well as KEGG databases to perform gene function and pathway annotation. MISA software was used to search microsatellite markers in the larval gut’s transcriptome. The specific primers of all SSRs were designed using Primer Premier 5 program and several pairs were used to amplify SSR loci in A. c. cerana larvae samples from 3 different regions (Beijing, Xingcheng, and Chengdu) in China by method of PCR. 【Result】 In this study, RNA seq produced 35 670 000 high quality reads, which were assembled into 43 557 unigenes with a mean length of 898 nt. 18 225 unigenes were annotated in the public protein databases. A total of 11 442 unigenes had a KOG classification and they distributed in 25 KOG categories, among them, RNA processing and modification was the largest group (1 249). 9 679 unigenes could be classified into three gene ontology (GO) categories, in which the mostly enriched ones were cellular process (4 201 unigenes), cell (2 900 unigenes) and binding (4 935 unigenes). 4 517 unigenes were annotated to 216 KEGG pathways, among them, ribosome (385 unigenes) was the largest. Finally, 13 448 SSRs were found in 7 763 unigenes, and 6 out 20 SSR loci could be successfully amplified in A. c. cerana larvae samples from 3 different regions in China using PCR. 【Conclusion】 This study assembled and annotated a reference transcriptome for A. c. cerana larval gut, which will provide a key information not only to studies on eastern honeybee and its larvae such as molecular biology and omics, but also to improve and validate the genome of A. cerana. SSR markers developed here could be applied to future investigation of A. c. cerana including gene map construction, genetic diversity analysis as well as gene location. Meanwhile, this study suggested that developing molecular markers using transcriptome data of non-model organism is a rapid and efficient method.
RNA seq; de novo assembly; Apis cerana cerana; unigenes; SSR
2016-09-29;接受日期:2016-12-12
国家现代农业产业技术体系(蜜蜂)建设专项(CARS-45-KXJ7)、福建省自然科学基金(2013J01070)
联系方式:徐细建,Tel:18020870542;E-mail:xxjlhj2006@163.com。郭睿,Tel:15205080780;E-mail:fafu_ruiguo@126.com。徐细建和郭睿为同等贡献作者。通信作者陈大福,Tel:0591-83726835;E-mail:dfchen826@163.com
猜你喜欢
信阳农林学院学报(2021年1期)2021-04-01
林业科技(2020年3期)2021-01-21
中国蜂业(2018年5期)2018-05-21
小天使·一年级语数英综合(2017年9期)2017-10-20
中国畜禽种业(2017年1期)2017-01-15
农村农业农民·B版(2016年7期)2016-10-21
阅读与作文(小学高年级版)(2016年5期)2016-05-10
阅读与作文(小学低年级版)(2016年1期)2016-03-12
小朋友·快乐手工(2015年11期)2016-01-07
中国蜂业(2014年8期)2014-01-26
