
类脂质蛋白沉积症两家系调查及ECM1基因突变检测
2017-04-07 02:42于文君付希安孙乐乐王真真张福仁
中国麻风皮肤病杂志 2017年3期
于文君 付希安 孙乐乐 王真真 刘 红 张福仁
·论著·
类脂质蛋白沉积症两家系调查及ECM1基因突变检测
于文君1,2付希安2孙乐乐2王真真2刘 红2张福仁2
目的: 检测类脂质蛋白沉积症二家系中细胞外基质蛋白(ECM1)基因突变位点。方法: 提取1号家系先证者及其母亲,2号家系先证者、父母、配偶及儿子外周血DNA。PCR技术扩增ECM1基因编码序列,采用一代Sanger法对PCR扩增产物进行测序。结果: 1号家系先证者在7号外显子发现已知突变(纯合突变c.960G>A),其母亲为杂合携带者;2号家系先证者为遗传复合体,是上述突变位点的杂合携带者,此外在3号外显子上存在1个插入突变c.142insC。结论: 类脂质蛋白沉积症存在遗传异质性。
类脂质蛋白沉积症; 细胞外基质蛋白1; 突变
类脂质蛋白沉积症(Lipoid proteinosis, LP),又称皮肤黏膜透明蛋白变性(hyalinosis cutis etmucosae)或Urbach-Wiethe病,是一种罕见的常染色体隐性遗传病。主要特点为玻璃样物质在各种组织中沉积引起的一系列临床症状,其中声音嘶哑多出现在婴儿时期[1];皮肤黏膜症状相对较晚,包括由轻微外伤或摩擦引起的口腔、面部、四肢的出血性血痂和水疱,分布于四肢伸侧(尤其是肘部)的疣状皮肤增生、蜡样丘疹[2],眼睑的串珠样丘疹(多个串珠丘疹沿着眼睑边缘和内眼角分布)等[3,4]。此外还常伴有皮肤以外的表现,如中枢神经系统病变和神经精神症状,如癫痫和行为异常及颞叶和海马钙化[5,6]。部分患者口腔、咽部、舌浸润可引起颌下腺炎症、舌系带缩短造成舌缩短难以伸出[7]。皮损组织病理:苏木紫伊红染色显示广泛的玻璃样物质沉积并破坏基底膜,对玻璃样物质进行染色,刚果红和PAS染色阳性但淀粉酶染色阴性[8],提示沉积物可能为糖蛋白和(或)蛋白聚糖。
该病普遍认为与细胞外基质蛋白1(Extracellular matrix protein 1,ECM1)缺乏有关。其编码基因ECM1位于1q21,至今已报道56例突变与LP相关(HGMD Professional 2016.3, http://www.hgmd.cf.ac.uk/ac/index.php)。本文对山东省2例LP家系进行基因检测。
1 材料与方法
1.1 临床资料 1号家系先证者,男,7岁。声音嘶哑7年伴双眼睑皮损1年。患者系第2胎,第2产,足月顺产,父母非近亲结婚,家族中无类似病例。体检:身高、智力发育正常,无明显的神经精神异常,无癫痫病史。皮肤科查体:双眼睑上缘见浅黄色透明状珍珠样丘疹,面部少量浅表凹陷性瘢痕,股沟处见暗红色浸润性斑片(图1a、b)。2号家系先证者为我院就诊的另一例类脂质蛋白沉积症患者,详细资料见文献[9]。
1.2 外周血DNA制备 经患者及其家属同意,分别抽取1号家系先证者及其母亲的外周血,2号家系先证者、父母、配偶及儿子外周血各5 mL于2% EDTA抗凝管中,使用QuickGene DNA whole blood kit L提取试剂盒提取基因组DNA。正常对照选取山东省皮肤病性病防治研究所中心实验室保存的100份山东省正常汉族人群DNA样本。
1.3 ECM1基因突变检测 针对ECM1基因10个外显子共合成8对引物[英潍捷基(上海)贸易有限公司],其中外显子2/3、6为自行设计,4/5按照文献[10],其余按照文献[1]合成。PCR采用25 μL体系,包括2xPCR Mix 12.5 μL,上下游引物(10 μM)各1 μL,基因组DNA(50 ng/μL)0.5 μL,ddH2O 10 μL。于基因扩增仪(ABI Veriti96 PCR)进行PCR反应。反应条件:94℃预变性10 min,之后共35个循环包括94℃变性30 s、60℃复性45 s、72℃延伸1 min。反应产物应用琼脂糖凝胶电泳进行产物验证。应用3130XL基因分析仪直接测序。
2 结果
对1号家系(图2a)2名成员进行检测,先证者(F1II1)在7号外显子存在一纯合同义突变c.960G>A,ENST00000369049(图2b)。既往实验发现该位点突变产生的潜在剪切受体位点可使ECM1基因发生剪切改变[11]。先证者母亲(F1I2)存在该位点的杂合突变(图2c)。
对2号家系(图3a)的5名成员PCR产物测序发现,先证者(F2II1)为遗传复合体,是1号家系突变位点的杂合携带者,即第960位点为G/A双峰,但是该突变并非遗传自其父母,而是自然产生;同时在3号外显子存在1个插入突变c.142insC(图3b),先证者母亲(F2I2)及儿子(F2III1)均为该突变杂合携带者(图3c)。在既往本院收集的8000份正常人外显子测序结果中进行对比未发现c.142insC突变。

图1 1a:1号家系先证者(F1II1)双眼睑上缘见浅黄色透明状珍珠样丘疹,面部少量浅表凹陷性瘢痕;1b:1号家系先证者股沟处暗红色浸润性斑片(箭头所示)
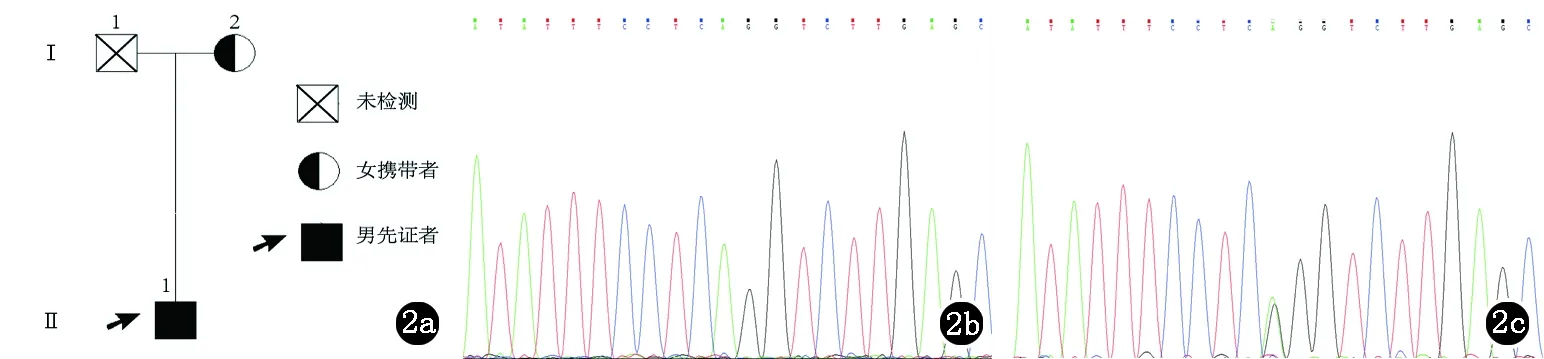
图2 2a:1号家系图;2b:1号家系先证者7号外显子c.960G>A纯合突变;2c:1号家系携带者7号外显子c.960G>A杂合突变

图3 3a:2号家系图;3b:2号家系3号外显子c.142insC插入突变;3c:2号家系7号外显子c.960G>A杂合突变
3 讨论
类脂质蛋白沉积症是一种罕见的常染色体隐性遗传病,其主要临床特点是声音嘶哑,皮肤、黏膜不同程度瘢痕形成,眼睑串珠样丘疹,颅内钙化等。至今已有超过300例LP报道[12]。Hamada等[13]在2002年通过基因连锁分析和候选对比等方法将其定位于ECM1基因,其编码的细胞外基质蛋白是一种分泌性糖蛋白,在表皮分化、真皮胶原和蛋白聚糖的结合、调节血管生成中具有重要生理作用[14]。ECM1基因发生突变可导致糖脂类或鞘脂类的降解异常,纤维胶原产量减少同时伴有基膜胶原的增多,ECM1蛋白和其他蛋白间结合力下降、细胞外基质稳定性下降,继而细胞间稳定性被削弱,最终造成皮肤或黏膜的易损。我们对山东省的两个LP家系进行基因分析,最终发现2处突变位点,其中7号外显子上的c.960G>A在两家系中均存在,该位点的杂合突变首次报道为1例山东LP患者[11],且证明该突变可导致ECM1基因的剪切发生改变,造成氨基酸序列变化。在1号家系先证者(F1II1)中首次发现该位点的纯合突变,其母(F1I2)为杂合携带,其父(F1I1)基因型未知。2号家系先证者(F2II1)为遗传复合体,是上述突变位点的杂合携带者,有趣的是该患者父母均不含有该突变,该位点为自然突变产生;此外,在3号外显子上存在1个插入突变c.142insC,造成移码突变,这是目前报道的第4例单碱基插入突变;家系中(F2III1)仅遗传了插入突变,提示2号家系先证者(F2II1)的两个突变位点位于不同染色体上。我们已根据情况为2号家系提供生育方面的遗传咨询,也将继续为其他患者或风险位点携带者提供遗传咨询。
[1] Hamada T, Wessagowit V, South AP, et al. Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation[J]. J Invest Dermatol,2003,120(3):345-350.
[2] Hamada T. Lipoid proteinosis[J]. Clin Exp Dermatol,2002,27(8):624-629.
[3] Urbach E, Wiethe C. Lipoidosis cutis et mucosae[J]. Virchows Arch,1929,273:285-319.
[4] Belliveau MJ, Alkhotani A, Ali A. Moniliform blepharosis of lipoid proteinosis[J]. JAMA Ophthalmol,2015,133(7):e150688.
[5] Siebert M, Markowitsch HJ, Bartel P. Amygdala, affect and cognition: evidence from 10 patients with Urbach-Wiethe disease[J]. Brain,2003,126(Pt 12):2627-2637.
[6] Thornton HB, Nel D, Thornton D, et al. The neuropsychiatry and neuropsychology of lipoid proteinosis[J]. J Neuropsychiatry Clin Neurosci,2008,20(1):86-92.
[7] Chan I, Liu L, Hamada T, et al. The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1[J]. Exp Dermatol,2007,16(11):881-890.
[8] Touart DM, Sau P. Cutaneous deposition diseases.Part I[J]. J Am Acad Dermatol,1998,39(2 Pt 1):149-171.
[9] 吴梅,陈明飞,周桂芝.类脂蛋白沉积症1例[J].中国麻风皮肤病杂志,2016,32(3):172-173.
[10] 王昌媛,章平肇,张福仁,等.类脂质蛋白沉积症一家系的基因突变检测[J].中华皮肤科杂志,2005,38(11):659-661.
[11] 高冬,连培文,陈剑,等.类脂蛋白沉积症一家系调查及基因突变检测[J].中华皮肤科杂志,2014,47(4):263-266.
[12] Mittal HC,Yadav S, Malik S, et al. Lipoid Proteinosis[J]. Int J Clin Pediatr Dent,2016,9(2):149-151.
[13] Hamada T, Mclean WH, Ramsay M, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1)[J]. Hum Mol Genet,2002,11(7):833-840.
[14] Chan I. The role of extracellular matrix protein 1 in human skin[J]. Clin Exp Dermatol,2004,29(1):52-56.
(收稿:2016-12-07 修回:2016-12-25)
Mutation analysis of ECM1 gene in two families with lipoid proteinosis
YUWenjun1,2,FUXi'an2,SUNLele2,WANGZhenzhen2,LIUHong2,ZHANGFuren2.
1.JinanUniversity,ShandongAcademyofMedicalSciences,Jinan250062,China; 2.ShandongProvincialInstituteofDermatologyandVenereology,Jinan250022,China
LIUHong,E-mail:hongyue2519@hotmail.com
Objective: To detect the mutations in the extracellular matrix protein 1 (ECM1) gene in two families with lipoid proteinosis (LP). Methods: Genomic DNA was extracted from peripheral blood of the propositus and the mother in family one and of the propositus, wife, parents and son in family two. The family numbers were all normal except propositus in two families. All the exons of ECM1 were amplified by PCR and the products were purified and directly sequenced to detect mutations by Sanger sequencing. Results: A homozygotic mutation of c.960G>A in exon 7 in the propositus and the heterozygous mutation of c.960G>A in the mather of family 1 was identified. One insertion mutation of c.142insC in exon 3 in the propositus and the heterozygous mutation of c.960G>A in the propositus of family 2 were identified. Conclusion: There exist genetic heterogenicity of LP.
lipoid proteinosis; extracellular matrix protein 1; mutation
山东省自科科学基金博士基金(编号:BS2013YY010)
1济南大学 山东省医学科学院 医学与生命科学学院,山东济南,250062
2山东省皮肤病性病防治研究所,山东济南,250022
刘红,E-mail:hongyue2519@hotmail.com
猜你喜欢
现代临床医学(2022年4期)2022-09-29
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
中老年保健(2021年12期)2021-08-24
种子(2021年3期)2021-04-12
中西医结合肝病杂志(2020年2期)2020-10-27
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
校园英语·下旬(2017年7期)2017-07-14
故事作文·高年级(2017年3期)2017-04-12
