
高效液相色谱法测定多西他赛原料药的含量
2017-03-09 07:37谷玉行王英利
湖北科技学院学报(医学版) 2017年1期
查 岭,谷玉行,王英利
(1.周口市中心医院,河南 周口 466000;2.上海现代哈森(商丘)药业有限公司)
高效液相色谱法测定多西他赛原料药的含量
查 岭1,谷玉行1,王英利2
(1.周口市中心医院,河南 周口 466000;2.上海现代哈森(商丘)药业有限公司)
目的 建立测定多西他赛原料药含量的方法。方法 色谱柱为Kromasil C18(5μm,250mm×4.60mm),流动相:甲醇-水(75∶25),流速:1.0mL/min,检测波长:232nm。结果 多西他赛原料药在高温、酸性和碱性条件下很不稳定,在氧化剂和光照条件下比较稳定,空白对照均无干扰;多西他赛浓度19.85~29.75μg/mL的范围内呈良好线性(r=0.9995),定量限为0.19μg/mL,检测限为0.052μg/mL,回收率在99.62%~100.06%之间,RSD为0.14%。结论 该方法操作简便、灵敏、准确度高,可用于多西他赛原料药的质量控制。
多西他赛原料药;含量测定;高效液相色谱
多西他赛化学名为{2aR-[2aα,4β,4aβ,6β,9α(αR*,βS*),11α,12α,12aα,12bα]}-β-{[(1,1-二甲基乙氧基)羰基]氨基}-α-羟基苯丙酸[12b-乙酰氧-12-苯甲酰氧-2a,3,4,4a,5,6,9,10,11,12,12a,12b-十二氢-4,6,11-三羟基-4a,8,13,13-四甲基-5-氧代-7,11-亚甲基-1H-环癸五烯并[3,4]苯并[1,2-b]氧杂丁环-9-基]酯三水合物,白色或类白色粉末,可以制成注射剂、脂质体[1]、干酏剂[2]等多种剂型,用于晚期乳腺癌、卵巢癌、非小细胞肺癌、胃癌等疾病的治疗[3]。为此,建立多西他赛原料药的含量的测定方法,控制其质量,在制剂方面有着重要意义。
1 实验材料
1.1 仪器 Agilent 1100型高效液相色谱仪,G1314A VWD型紫外检测器,Agilent Chem Station 工作站,Sartorius BP211D型电子天平。
1.2 药品与试剂 多西他赛原料药(批号为20140501,20140502,20140503,湖北信康医药化工有限公司);多西他赛对照品(中国食品药品检定研究院,100666-201408);甲醇为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件 色谱柱Kromasil C18柱(5μm,250mm×4.60mm);流动相:甲醇-水(75∶25);流速:1.0mL/min;柱温:30℃;检测波长:232nm;进样量:20μL。
2.2 溶液制备
2.2.1 供试品溶液 取25mg多西他赛,精密称定,置于25mL容量瓶中,用流动相溶解并稀释至刻度;精密量取5.0mL上述溶液置于50mL容量瓶中,用流动相稀释至刻度,摇匀。
2.2.2 对照品溶液 取25mg多西他赛对照品,精密称定,置于25mL容量瓶中,用流动相溶解并稀释至刻度;精密量取5.0mL上述溶液置于50mL容量瓶中,用流动相稀释至刻度,摇匀。
2.3 系统适用性和专属性试验
2.3.1 系统适用性 取2.2.2项下的对照品溶液和供试品溶液按2.1项下色谱条件进样测定,记录色谱图(详见图1);进样5次主峰的理论板数最小为8178.8,主峰面积的RSD为0.52%,保留时间的RSD为0.59%,符合要求。
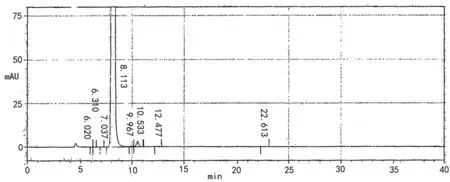
A 供试品

B对照品
2.3.2 专属性试验 取多西他赛原料药25mg,分别置于25mL量瓶中,按下列方法进行破坏试验。①酸破坏:取本品,加1mol/L的盐酸溶液5mL,室温放置4h后,加入等量的1mol/L的氢氧化钠溶液中和,再加流动相稀释至刻度,摇匀。②碱破坏:取本品,加入1mol/L的氢氧化钠溶液5mL,室温放置4h后,加入等量的1mol/L的盐酸溶液中和,再加流动相稀释至刻度,摇匀。③高温破坏:取多西他赛在150℃干燥4h,再取约25mg置25mL量瓶中,加流动相使溶解,并稀释至刻度,摇匀。④光照破坏:取本品,加流动相使溶解并稀释至刻度,置4000lx光照下4h,摇匀。⑤氧化破坏:取本品,加入30%双氧水5mL,室温放置4h后,再加流动相使完全溶解并稀释至刻度,摇匀。⑥空白1(双氧水):取30%双氧水5mL,至25mL量瓶中,加流动相稀释至刻度。⑦空白2(酸碱中和液):取1mol/L的盐酸溶液和1mol/L的氢氧化钠溶液各5mL,置25mL量瓶中,加流动相稀释至刻度,摇匀。
结果表明多西他赛原料药在高温、酸性和碱性条件下很不稳定,在氧化剂和光照条件下比较稳定,空白对照均无干扰。
2.4 线性关系考察 取多西他赛对照品25mg,精密称定,置25mL量瓶中加流动相使溶解并稀释至刻度作为对照品贮备溶液。再精密量取贮备液0.8mL、0.9mL、1.0mL、1.1mL、1.2mL分别置10mL量瓶中,加流动相稀释至刻度。制成80μg/mL、90μg/mL、100μg/mL、110μg/mL、120μg/mL的溶液,作为供试品系溶液。取各供试品系列溶液,在含量的色谱条件下,分别进样一次,记录色谱图。以溶液浓度为横坐标(X),峰面积为纵坐标(Y)作图,得回归方程y=90276x+62820,多西他赛浓度19.85~29.75μg/mL的范围内呈良好线性。(线性范围:由进样浓度的80%~120%),在此范围内线性相关系数R为0.9995。
2.5 检测限与定量限 取多西他赛对照品溶液,逐步稀释后进样,记录色谱图,使多西他赛主峰为基线噪音的10倍左右,计算出定量限为0.19μg/mL。将最小定量限的溶液继续稀释后进样,记录色谱图,使多西他赛主峰为基线噪音的3倍左右,计算出检测限为0.052μg/mL。
2.6 精密度(重复性)试验 配制6份供试品溶液及2份对照品溶液。按照含量色谱条件对照品溶液1×5,对照品溶液2×2,各供试品溶液×2,记录色谱图,计算各供试品溶液的含量。对照溶液五针峰面积RSD为0.54%,测定6次结果的RSD为0.18%,表明本实验有良好的精密度和重复性。
2.7 稳定性试验 取多西他赛对照品溶液,室温条件下,于0、2、4、8、12、24、48及72h,在含量色谱条件下,分别进样,记录色谱图。峰面积的RSD为1.6%,证明对照溶液配制后,72h内使用,对多西他赛含量的测试结果没有影响。
2.8 回收率试验 精密称取多西他赛原料药20mg、25mg、30mg,分别置于25mL容量瓶中,用流动相溶解并稀释至刻度(必要时超声助溶);分别精密量取1mL上述溶液置于10mL容量瓶中,用流动相稀释至刻度,摇匀(各制备该溶液3份)作为供试品溶液。另取25mg多西他赛对照品,精密称定,置于25mL容量瓶中,用流动相溶解并稀释至刻度(必要时超声助溶);精密量取1mL上述溶液置于10mL容量瓶中,用流动相稀释至刻度,摇匀。对照溶液五针峰面积RSD为0.54%,多西他赛9次测得的回收率在99.62%~100.06%之间,9份溶液回收率的RSD为0.14%。
2.9 溶液稳定性试验 取多西他赛对照品溶液,室温条件下,于0、2、4、8、12、24、48及72h,在含量色谱条件下,分别进样,记录色谱图。多西他赛含量溶液放置72h,峰面积的RSD为1.6%,表明对照溶液配制后,72h内使用,对多西他赛含量的测试结果没有影响。
2.10 原料药多西他赛的含量测定 分别取对照品溶液和供试品溶液进行测定,记录色谱图,按外标法以峰面积计算3批的含量分别为98.6%,99.2%和98.8%。
有关物质:取样品,精密称定,用流动相溶解并稀释制成每1mL中含1mg的溶液,作为供试品溶液;精密量取适量,用流动相稀释制成每1mL中含10μg的溶液,作为对照溶液。照含量测定项下的色谱条件,量取对照溶液20μL,注入高效液相色谱仪,调节仪器灵敏度,使主成分色谱峰高约为满量程的10%~20%;再量取供试品溶液与对照溶液各20μL,分别注入高效液相色谱仪,记录色谱图至主成分峰保留时间的3倍,结果3批最大单个杂质含量分别为0.13%、0.21%和0.14%;总杂质含量分别为0.51%、0.26%和0.17%。
3 讨 论
多西他赛是紫杉烷类抗肿瘤药,目前临床上和表柔比星、卡铂、奈达铂、塞替哌等药物制成多个有效的化疗方案,临床使用非常广泛,除了注射剂外,其他剂型如脂质体、胶束、微球等剂型也都正在研发之中,金龙宇等[4]和李婧等[5]建立了注射用多西他赛的高效液相测定方法,本实验对多西他赛原料药的质量控制进行了探讨。
在其它色谱条件不变情况下,分别在不同波长(230nm、232nm、234nm),依次进样:空白溶剂、供试品溶液、对照溶液。多西他赛与相邻峰之间的最小分离度为3.23、3.29和3.49主峰的理论板数8437、8514和8394,空白溶剂无干扰,检测波长在230~234nm,能够满足有关物质系统适用性要求,耐用性良好。
流动相在甲醇-水比例(70∶30)~(75∶25),能够满足有关物质系统适用性要求;流动相甲醇-水(80∶20)在主峰与6.770min的杂质峰之间有一杂质包夹至6.770min杂质上,无法检出,不能用于检测。
[1]付盟,龚伟,张慧,等.多西他赛热敏脂质体的制备及含量和包封率的测定[J].军事医学,2012,36(3):196
[2]董文凤,李波,徐希明,等.多西他赛干酏剂的研制[J].中国医药工业杂志,2014,45(3):240
[3]陈新谦.新编药物学[M].第17版.北京:人民卫生出版社,2011:751
[4]金龙宇,邱利焱,金一,等.注射用多西他赛的HPLC测定[J].中国医药工业杂志,2006,37(10):708
[5]李婧,黄毅岚,张丹,等.HPLC测定注射用多西他赛的含量并检查有关物质[J].华西药学杂志,2007,22(6):692
Determination of Docetaxel Drug Substance by HPLC
CHA Ling,GU Yu-hang,WANG Ying-li
(TheCentralHospitalofZhoukou,ZhoukouHenan466000,China)
Objective To establish a method for determination of docetaxel drug substances. Methods The analysis was performed on Kromasil C18 column(250mm×4.6mm,5μm) with detective wave length at 232nm.The mobile phase consisted of a mixture of methanol and water(75∶25) with the flow rate of 1.0ML/min.Results Docetaxel drug substance is not stable in the high temperature,acidic and alkaline conditions,but is relatively stable in the oxidant and light conditions.There is no interference in the blank control.Docetaxel concentration of 19.85μg/mL to 29.75μg/mL showed good linearity(r=0.9995).The limit of quantification was 0.19μg/mL and the detection limit is 0.052μg/mL.The recovery rate was 99.62%~100.06% and RSD 0.14%.Conclusion This method is simple,sensitive,accurate and available for quality control of docetaxel drug substance.
Docetaxel drug substane;Content determination;HPLC
R927.2
A
2095-4646(2017)01-0003-03
10.16751/j.cnki.2095-4646.2017.01.0003
2016-11-29)
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
中国盐业(2018年20期)2019-01-14
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
中国当代医药(2015年33期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
天津药学(2013年2期)2013-12-23
