
改进的Leuckart-Wallach反应合成4,4-二甲基环己胺
2017-03-08 06:18尹大力
化学研究 2017年1期
杨 旭,张 翔,尹大力
(中国医学科学院 北京协和医学院药物研究所,“活性物质发现与适药化研究”北京市重点实验室,北京 100050)
改进的Leuckart-Wallach反应合成4,4-二甲基环己胺
杨 旭,张 翔*,尹大力
(中国医学科学院 北京协和医学院药物研究所,“活性物质发现与适药化研究”北京市重点实验室,北京 100050)
在综述现有烷基取代环己胺合成方法的基础上,以4,4-二甲基环己胺为例介绍一种优化改进的Leuckart-Wallach反应.优化后的反应以甲醇/氨水9∶1为溶剂体系,10%钯/碳为催化剂,利用甲酸铵为氢源和氮源,一锅法还原胺化4,4-二甲基环己酮得到4,4-二甲基环己胺,该方法产品收率和纯度高,条件温和,操作简便,适用于工业化生产.
4,4-二甲基环己胺;甲醇/氨水;甲酸铵;还原胺化
烷基取代环己胺是常用的有机合成砌块,同时也是医药工业中重要的合成原料,许多已上市或在研的药物分子中均包含此类结构单元.例如,磺酰脲类抗糖尿病药格列美脲(Glimepiride, 1)[1]、新型阿片类受体拮抗剂2和3[2]、诺华公司的两个处于临床前研究的抗TB候选药物NITD-304和NITD-349[3]等(图1).因此,开发适用于工业化生产的烷基取代环己胺的合成方法是很有必要的.
目前,常用的合成烷基取代环己胺的方法是烷基取代环己酮与盐酸羟胺反应形成肟,然后利用还原剂如四氢铝锂[4]、兰尼镍[5]、无定形镍粉和硼氢化钠[6]等还原肟得到产物;或者烷基取代环己酮与苯甲醛反应形成亚胺,硼氢化钠还原亚胺得到N-苄基取代的环己胺,然后再脱除苄基得到产物[7](图2).但是两种策略均存在显著缺欠:路线(1)中四氢铝锂对水很敏感,且后处理时很容易形成胶状物导致操作困难、产物收率低,而兰尼镍还原需高压条件,经济成本较高,存在安全隐患;路线(2)的原子经济性较差,且同样需较高压力下氢化或贵金属催化.上述的各种传统方法均不适用于工业化生产.
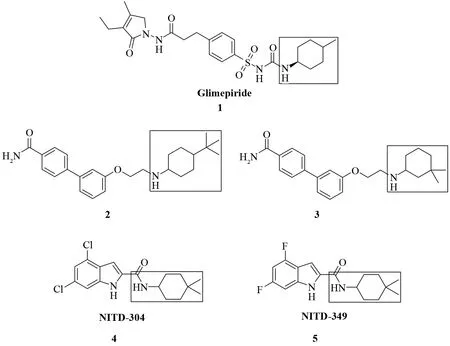
图1 含有烷基取代环己胺结构片段的化合物Fig.1 Compounds containing alkyl substituted cyclohexan-1-amines
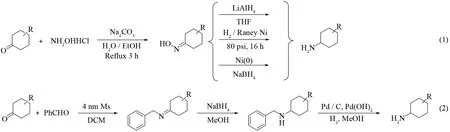
图2 烷基取代环己胺的常用合成方法Fig.2 Common methods for synthesis of alkyl substituted cyclohexan-1-amines
经典的Leuckart-Wallach反应[8-9]利用甲酸铵[10]作为氢源和氮源可以从酮一步还原胺化得到相应的胺,但反应需要在高温条件下进行,条件苛刻,且副产物较多.ALLEGRETTI等[11-13]报道对该方法进行改进,加入钯/碳作为催化剂,甲醇/水9∶1体系作为混合溶剂,可以在室温条件下一锅法还原胺化羰基化合物得到相应的胺,反应条件温和.
但这种改进的Leuckart-Wallach反应无法避免仲胺副产物的明显生成,降低了目标化合物的产率,也给最终产物的分离纯化带来很大的困难,因此该改进方法的实用性依然受到制约.为实现实用性目标,本文将其用于4,4-二甲基环己胺的合成时,对反应条件进一步优化,在溶剂系统中应用氨水代替水,以此抑制仲胺副产物的生成[14](图3),取得了较理想的结果.关于使用甲醇/氨水9∶1混合溶剂体系,在钯/碳催化的还原胺化制备伯胺反应中用以抑制副产物仲胺生成的方法,至今尚未发现有相关文献报道.
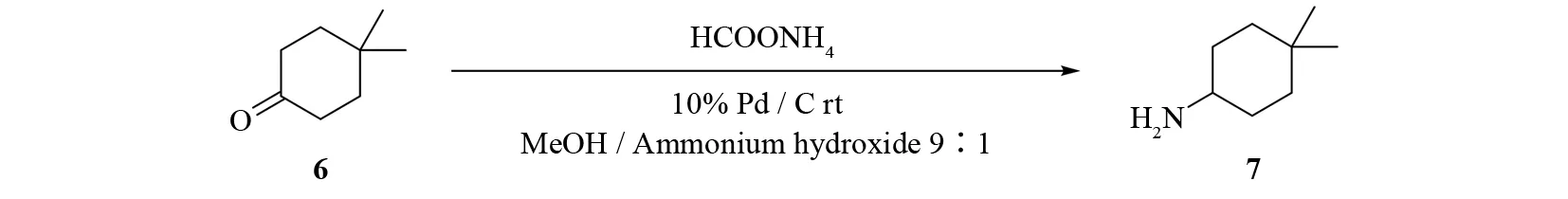
图3 钯/碳催化的还原胺化法制备4,4-二甲基环己胺Fig.3 Synthesis of 4,4-dimethylcyclohexan-1-amine by reductive amination with Pd/C as the catalyst
1 实验部分
1.1 仪器与试剂
AVANCEIII400型核磁共振仪(瑞士Bruker公司);Thermo Exactive Orbitrap plus spectrometer (ESI-MS)型质谱仪(美国赛默飞公司);GC-2010 plus型气相色谱仪(日本岛津公司);MP-J3型熔点仪(日本Yanaco公司).
4,4-二甲基环己酮(96%,北京偶合科技有限公司);10%钯/碳(试剂级,萨恩化学技术(上海)有限公司);甲酸铵(分析纯,天津市大茂化学试剂厂);25%~28%氨水(分析纯,北京百灵威科技有限公司);其他试剂溶剂均为常规分析纯.
1.2 化合物的合成
取450 g(3.57 mol)4,4-二甲基环己酮置于20 L玻璃反应釜中,依次加入2.25 kg(35.66 mol)甲酸铵,6.7 L甲醇,740 mL氨水,搅拌至完全溶解,加入225 g 10%钯/碳,室温搅拌,TLC跟踪反应进程,3 h后反应结束.过滤,滤去钯/碳,减压蒸干滤液,加入6.7 L H2O溶解残留物,缓慢向水相中加入氢氧化钠固体,调pH至13~14之间,二氯甲烷(2.5 L × 3)萃取三次,合并有机相,无水硫酸钠干燥有机相.过滤,减压蒸干溶剂,得浅黄色油状物,加异丙醇溶解,搅拌的条件下缓慢向其中滴加浓盐酸,直至pH到1~2.减压蒸干溶剂,得黄色固体.然后加入异丙醇,外温80 ℃加热回流30 min后加入叔丁基甲基醚,降至室温,抽滤,滤饼用叔丁基甲基醚洗,50 ℃鼓风干燥,得白色固体421 g,收率72.6%,m.p.>250 ℃.1H NMR(DMSO-d6,400MHz)δ:8.24(br s,2H); 2.85(m,1H); 1.76~1.72(m,2H); 1.60~1.50(m,2H); 1.39~1.35(m,2H); 1.22~1.15(m,2H); 0.88(s,6H ).13C NMR(CDCl3,100MHz)δ:24.79; 27.54; 29.47; 31.58; 36.91; 50.99.HRMS(ESI):m/z[M+H]+Calcd.For C6H18N:128.143 38,Found 128.143 72.
1.3 气相色谱测试方法
RESTEK色谱柱,型号:Rtx-5,规格:30 m*0.25 mm,0.25 μm.
1.3.1 对照品溶液的配制
精密称取一级胺及二级胺对照品(自制)约12 mg,置50 mL量瓶中,加二氯甲烷溶解并稀释至刻度,摇匀,取上述溶液2 mL,加0.5 mol/L氢氧化钠溶液1 mL混合均匀,取二氯甲烷层,作为对照品溶液.
1.3.2 供试品溶液的配制
取样品约20 mg,置10 mL量瓶中,加甲醇适量溶解,用二氯甲烷稀释至刻度,摇匀,作为供试品溶液.
1.3.3 色谱条件
以5%二苯基-95%二甲基聚硅氧烷为固定液(或极性相近的固定液)的毛细管柱为色谱柱;起始温度为80 ℃,以15 ℃/min的速率升温至300 ℃,维持10 min;检测器为FID,检测器温度310 ℃;进样口温度300 ℃;载气为氮气,流速为2 mL/min;分流比5∶1.
1.3.4 测定法
分别精密量取供试品溶液和对照品溶液各1 μL注入气相色谱仪,记录色谱图,按外标法以峰面积计算一级胺及二级胺的含量.
2 实验结果与讨论
2.1 甲醇/氨水9∶1溶剂系统对副产物的抑制作用及机理
在以甲醇/水9∶1体系作为溶剂系统的反应过程中,我们发现有相应的仲胺副产物8生成.由于仲胺与主产物的相似性,在后处理过程中无法通过萃取、洗涤等简单操作去除,只能通过高真空蒸馏或者柱层析来分离目标化合物和仲胺,这使得目标产物的收率偏低,在40%左右.
根据经典Leuckart反应的机理,推测产生仲胺的过程如下(图4).4,4-二甲基环己酮先与甲酸铵分解得到的氨反应生成相应的亚胺9,亚胺性质很活泼,既可以加氢还原得到相应的伯胺即目标化合物7,也可以与伯胺反应得到1-氨基二烷基胺11,随即放出氨转化为席夫碱12形成平衡反应,然后席夫碱经与9类似的过程加氢还原得到仲胺8.
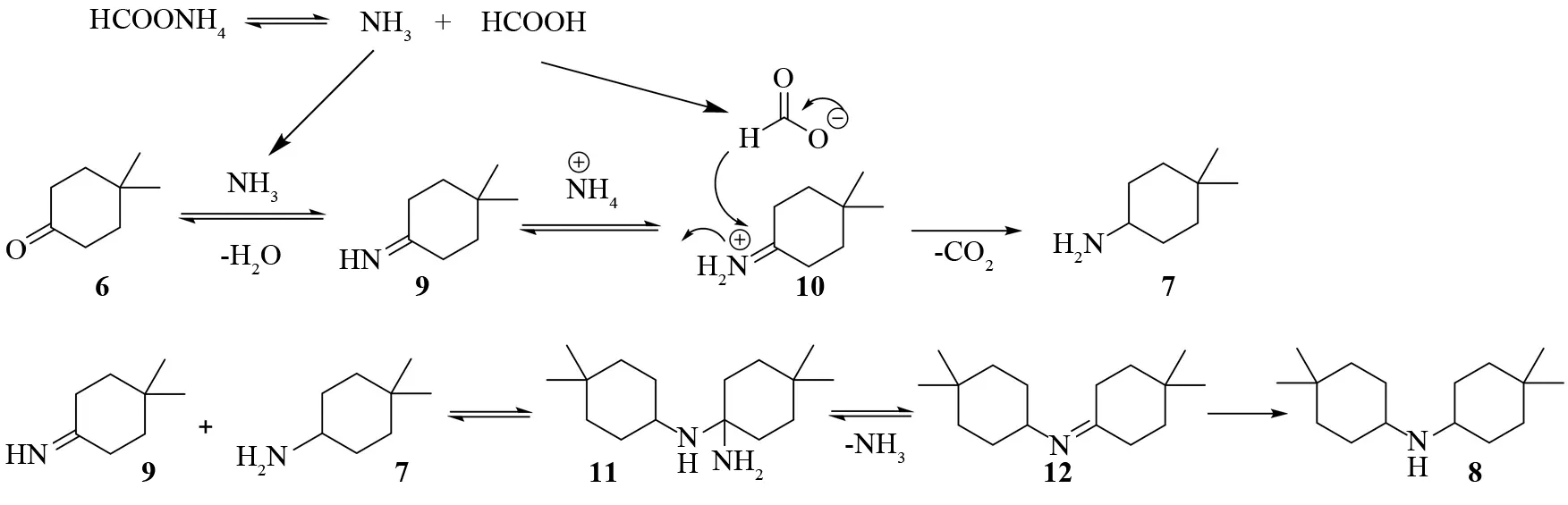
图4 甲醇/水9∶1体系还原胺化法制备4,4-二甲基环己胺的反应机理Fig.4 Reaction mechanism of synthesis of 4,4-dimethylcyclohexan-1-amine by reductive amination with methanol/water (9∶1) as the solvents
基于上述推测,我们考虑加入氨水,应该可以抑制仲胺的产生,可能的反应机理如图5所示.亚胺9与氨水形成13和14,13加氢还原得到相应伯胺,14失去水又重新生成亚胺9.氨水的存在竞争性抑制亚胺与伯胺的反应,从而抑制副产物仲胺8的生成.在用25%~28%氨水代替水与甲醇组成甲醇/氨水9∶1的混合溶剂体系后,TLC监测显示反应体系中基本无仲胺8的生成,气相色谱显示粗产物中伯胺与仲胺的比例由25∶1升高至78∶1,使纯化后目标产物的收率可提高到70%左右的水平.
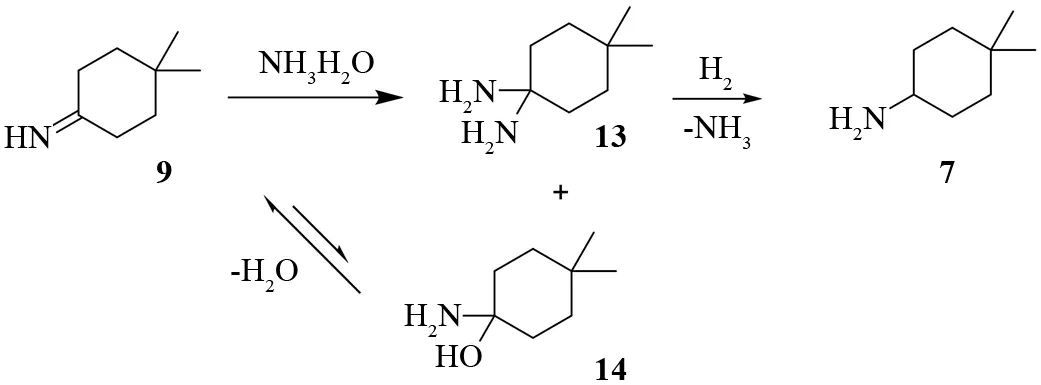
图5 氨水抑制仲胺形成的机理Fig.5 Mechanism of ammonium hydroxide inhibiting the formation of secondary amines
2.2 Pd/C使用量对反应的影响
Pd/C在还原胺化反应中促进甲酸铵分解为氨和甲酸,同时催化亚胺的加氢还原.当甲酸铵与4,4-二甲基环己胺的物质的量之比为10∶1、溶剂系统为甲醇/氨水9∶1时,在常温常压条件下,本文考察了Pd/C与底物4,4-二甲基环己酮的质量比对反应的影响,见表1.

表1 Pd/C使用量对反应的影响Table 1 Effect of Pd/C amount on the reaction
在实验中,随着Pd使用量的降低,TLC监测反应时间明显延长.且Pd/C的质量比为1∶10时,杂质明显增多,目标化合物相对含量明显减少;而Pd/C质量比为1∶2时,TLC显示其反应结果与Pd/C质量比为1∶1时无明显差异,因此最终选择Pd/C质量比1∶2作为最优的反应条件.
2.3 Pd/C循环使用次数对反应的影响
由于实验中Pd/C的使用量比较大,因此在保持其他实验条件不变的情况下本文考察了Pd/C循环使用的次数对反应的影响.由表2可知,Pd/C循环使用7次,反应均可在3 h内迅速完成,TLC监测显示反应结果无明显差异;而第八次时Pd/C活性有明显降低,反应时间延长至10 h,TLC显示杂质增多.尽管该反应中的Pd/C单次用量较大,但可循环使用七次,同时Pd/C可回收再生,因此仍具有成本优势.

表2 Pd/C循环使用次数对反应的影响Table 2 Effect of Pd/C recycle times on the reaction
2.4 后处理方法对反应收率的影响
胺的常规纯化方法是与盐酸成盐,然后利用醇类溶剂重结晶得到相应的盐酸盐产物.产物盐酸盐在醇类溶剂中溶解度较高,重结晶的收率低于10%.对纯化过程优化,将粗品油状物成盐后,用异丙醇和叔丁基甲基醚的混合溶剂打浆处理,过滤可得纯品,收率提高至70%左右.
3 结论
以甲醇/氨水9∶1为溶剂体系,钯/碳为催化剂,利用甲酸铵作为氢源和氮源,一锅法还原胺化4,4-二甲基环己酮得到4,4-二甲基环己胺,其中氨水可有效抑制还原胺化反应过程中副产物仲胺的形成,钯/碳可循环使用多次,经济成本较低,该方法可应用于工业化生产.
[1] KADAM S,TARUR V,NAIK S,et al.Process for preparation of substantially pure glimepiride:US,156343 [P].2007-04-12.
[2] IGNAR D M.Method of treatment using novel antagonists or inverse agonists at opioid receptors:US,608463 [P].2010-05-06.
[3] RAO S P,LAKSHMINARAYANA S B,KONDREDDI R R,et al.Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis [J].Science Translational Medicine,2013,5(214):214ra168.
[4] WANG J,MA C,WU Y,et al.Exploring organosilane amines as potent inhibitors and structural probes of influenza a virus M2 proton channel [J].Journal of the American Chemical Society,2011,133(35):13844-13847.
[5] JIRICEK J,KONDREDDI R R,SMITH P W.Indole carboxamide derivatives and uses thereof:US,420074 [P].2015-06-25.
[6] LIU S,YANG Y,ZHEN X,et al.Enhanced reduction of C-N multiple bonds using sodium borohydride and an amorphous nickel catalyst [J].Organic & Biomolecular Chemistry,2012,10(3):663-670.
[7] BANDAR J S,SAUER G S,WULFF W D,et al.Transition state analysis of enantioselective brønsted base catalysis by chiral cyclopropenimines [J].Journal of the American Chemical Society,2014,136(30):10700-10707.
[8] CHUBB F,HAY A S,SANDIN R B.The Leuckart reaction of some 1,5-diketones [J].Journal of the American Chemical Society,1953,75(23):6042-6044.
[9] MOORE M L.Organic reactions Vol 5 [M].New York:John Wiley & Sons Inc,1949:301-330.
[10] 孙利民,孟昭力,吉爱国.甲酸铵在催化转移氢化反应中的应用[J].化学试剂,2005,27(5):279-282.
SUN L M,MENG Z L,JI A G.Application of ammonium formate in catalytic,transter hydrogenation [J].Chemical Reagents,2005,27(5):279-282.
[11] ALLEGRETTI M,BERDINI V,CESTA M C,et al.One-pot,new stereoselective synthesis of endo-tropanamine [J].Tetrahedron Letters,2001,42(25):4257-4259.
[12] ALLEGRETTI M,ANACARDIO R,CESTA E A.A practical synthesis of 7-azaindolylcarboxy-e ndo-tropanamide (DF 1012) [J].Organic Process Research & Development,2003,7(2):209-213.
[13] BERDINI V,CESTA M C,CURTI R,et al.A modified palladium catalysed reductive amination procedure [J].Tetrahedron Letters,2002,58(28):5669-5674.
[14] GREENFIELD H.Catalytic hydrogenation of butyronitrile [J].Industrial & Engineering Chemistry Product Research and Development,1967,6(2):142-144.
[责任编辑:张普玉]
Synthesis of 4,4-dimethylcyclohexan-1-amine by improved Leuckart-Wallach reaction
YANG Xu,ZHANG Xiang*,YIN Dali
(InstituteofMateriaMedica,ChineseAcademyofMedicalScience&PekingUnionMedicalCollege,BeijingKeyLaboratoryofActiveSubstanceDiscoveryandDruggabilityEvaluation,Beijing100050,China)
A method for the synthesis of alkyl substituted cyclohexan-1-amines was summarized.A new condition for reductive amination of 4,4-dimethylcyclohexan-1-one to form 4,4-dimethylcyclohexan-1-amine by improved Leuckart-Wallach reaction was described.With methanol/ammonium hydroxide (9∶1) as the solvents and palladium/carbon as a catalyst,the reaction was carried out by using 4,4-dimethylcyclohexan-1-one as a substrate,ammonium formate as a source of nitrogen and hydrogen.The condition of this one-pot reaction was mild with convenient work-up.The yield of target product was higher and the purity of it was better.Accordingly,this method is suitable for industrial production.
4,4-dimethylcyclohexan-1-amine; methanol/ammonium hydroxide; ammonium formate; reductive amination
2016-11-05.
国家科技重大专项“重大新药创制”(2015ZX09102007-004),中国医学科学院医学与健康科技创新工程(2016-I2M-3-022).
杨 旭(1992-),男,硕士生,研究方向为药物小分子的设计与合成.*
, E-mail:simon@imm.ac.cn.
O624.6
A
1008-1011(2017)01-0039-05
猜你喜欢
化学反应工程与工艺(2022年1期)2022-04-23
中学生理科应试(2021年10期)2021-12-07
化工管理(2021年7期)2021-05-13
上海计量测试(2020年1期)2020-03-18
化学反应工程与工艺(2017年6期)2017-06-12
制冷技术(2016年2期)2016-12-01
浙江大学学报(工学版)(2016年2期)2016-06-05
药学研究(2015年11期)2015-12-19
郑州大学学报(工学版)(2015年6期)2015-03-18
中国洗涤用品工业(2012年4期)2012-03-20
