
DFT方法研究基于脲及硫脲衍生物的阴离子受体
2017-03-08 06:18:41耿玮宏王亚茹张瑞红王炳强
化学研究 2017年1期
耿玮宏,王亚茹,李 夏,张瑞红,王炳强
(山西师范大学 化学与材料科学学院,山西 临汾 041000)
DFT方法研究基于脲及硫脲衍生物的阴离子受体
耿玮宏,王亚茹,李 夏,张瑞红,王炳强*
(山西师范大学 化学与材料科学学院,山西 临汾 041000)
采用密度泛函理论(DFT)模拟了两个基于脲和硫脲衍生物的受体分子对卤素阴离子的识别过程.结构优化表明基于脲衍生物的受体分子1最稳定构象为“反反”构象,分子内部形成稳定的Cα-H…O=C分子内氢键;而基于硫脲衍生物的受体分子2,不能形成分子内氢键,最稳定构象为“反顺”构象.受体1、2与卤素阴离子F-、Cl-可形成稳定的双氢键复合物,在此过程中,受体2经历了由“反顺”构象到“反反”构象的异构化过程.结构和能量分析表明,1、2受体分子与F-离子间的氢键强度远大于其与Cl-离子间的氢键;另一方面,受体2与阴离子间的氢键明显强于受体1,这是由于硫脲基N-H键具有更强的酸性.此外,对受体分子、氢键复合物及去质子化产物的吸收光谱计算结果表明,受体与F-离子作用可产生明显的吸收光谱红移,而与Cl-离子的作用对光谱影响较小.
脲;硫脲;阴离子受体;密度泛函理论
随着阴离子在催化化学、环境化学、医药与生命科学等领域所扮演的角色愈加重要,对阴离子的检测、分离和识别显得更具研究和应用价值.因此,设计并合成对特定阴离子具有高亲和性、高选择性的受体已经成为当前的研究热点[1-2].受体与阴离子之间的识别主要是通过非共价键作用如氢键、疏水作用、静电作用及Lewis酸中心配位作用等进行.其中氢键因具有很好的方向性、选择性,且容易形成,而被作为阴离子受体设计与合成中应用最广泛的一种作用力[3].酰胺[4]、多胺[5]、脲[6]、硫脲[7]、胍[8]和酚羟基[9]等都可作为氢键的供体,与阴离子形成氢键复合物.而脲和硫脲因可以提供双质子与阴离子形成双氢键,而常被用作阴离子识别的中性受体[6-7].
除了大量的实验研究外,理论计算也在阴离子受体设计方面发挥了极其重要的作用.如GHOSH等[10]用密度泛函理论(DFT)的方法模拟在尿素两端分别引入不同的吸电子和供电子基团,讨论取代基效应对阴离子的识别过程的影响,结果表明可以通过调节N-H单元的酸性,从而实现对不同阴离子的选择性识别.而LI等[11]则通过DFT方法对2-(2′-苯基脲苯)苯丙恶唑脲对氟离子的感应机理进行了研究,结果表明在此受体分子存在着激发态分子内的质子转移(ESIPT),加入氟离子之后可以捕获其N-H上的质子形成氢键,从而实现对该受体的识别.作者还计算了识别体系的紫外可见光谱及荧光光谱,结果和实验数据相近,说明理论计算不但可以从结构方面还可从电子行为的角度来探讨识别机理.
最近,GOMEZ等[12]合成了新的基于脲及硫脲衍生物的阴离子受体(1:1-(2-甲基-1,3-二氧代异吲哚-5-基)-3-苯基脲;2:1-(2-甲基-1,3-二氧代异吲哚-5-基)-3-苯基硫脲),对比分析了这些受体分子对羧酸根、卤离子、磷酸根等无机阴离子的识别.在本文中,我们使用DFT方法,对这两个阴离子受体分子进行计算研究.我们计算了两个受体分子的4种(反顺、顺反、反反、顺顺)稳定构象(如图1所示),讨论了两种受体与氟、氯阴离子的氢键相互作用、氢键及去质子化过程对受体分子吸收光谱的影响,为进一步设计合成更高效的基于脲或硫脲基的受体提供理论依据.
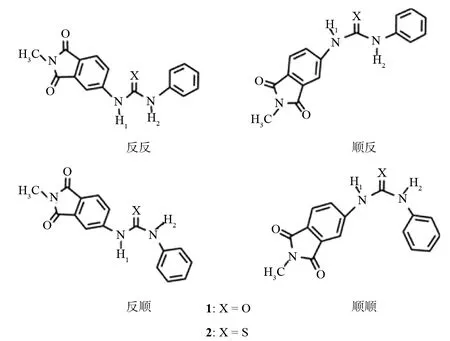
图1 受体1、2的4种稳定构象Fig.1 Four stable conformers of the receptors 1 and 2
1 计算方法
在B3LYP/6-311++G(d,p)水平上,对脲及硫脲基阴离子受体1、2,其与卤素阴离子F-、Cl-的氢键复合物,及受体分子去质子化产物的几何结构进行优化.在相同水平上计算优化结构的谐振频率,所有频率值均为正值,表明所得稳定结构均为势能面上的极小值点.使用MP2方法及相同基组,计算了4种氢键复合物的相互作用能,并使用完全均衡校正法(Counterpoise,CP)校正了计算中的基组重叠误差(BSSE).此外,分别使用TD-B3LYP和TD-CAM-B3LYP[13-14]方法计算了受体1、2与卤素离子的复合物及去质子化产物的吸收光谱.两种方法得到光谱数值相差较大,说明长程校正在光谱计算中非常重要[15].因此在下述讨论中,使用CAM-B3LYP/6-311++G(d,p)方法所得的光谱数据.所有计算使用DMSO溶剂,通过极化连续介质模型(PCM)模拟了溶剂化效应.所有计算均通过GAUSSIAN 09程序[16]完成.
2 结果与讨论
2.1 分子几何构型的分析
使用B3LYP/6-311++G(d,p)方法,对1和2两个受体分子的稳定构象进行几何优化,并在MP2水平上计算了这些构象的能量.表1列出了受体1、2的4种构象的一些关键键参数(原子编号见图2).对于基于脲基的受体1,“反反”构象是最稳定的,“反顺”、“顺反”、“顺顺”构象的能量依次高出4.35、10.14、23.81 kJ·mol-1.但是对于基于硫脲的受体2,“反顺”构象则最稳定,“顺反”、“顺顺”、“反反”构象的能量依次比其高出3.62、、9.45 kJ·mol-1.
对于受体1的“反反”构象,脲基与异吲哚环及苯环基本共面,二面角DC1-N1-C2-C4和DC1-N2-C3-C5均为180.0°.在“反反”构象中,羰基氧距两侧芳环氢原子的距离分别为0.218 2 和0.222 3 nm,分子内形成两个Cα-H…O=C(Cα表示芳环上的C原子)型氢键,这是受体1能够保持平面结构的主要原因.其他3种构象则偏离平面结构形成了一定夹角.在受体1中,4种构象中N-H键键长均接近0.101 0 nm,说明构象的变化对N-H键几乎没有影响.但对其他键长键角则产生了一些影响,如对于左侧的异吲哚环来说,处于顺式位置(“顺反”、“顺顺”构象)时比相应反式位置(“反反”、“反顺”构象)的RC2-N1长0.000 7 nm,而键角AC2-N1-C1要大2.3° ~3.0°;而对于右侧的苯环来说,处于顺式位置(“反顺”、“顺顺”构象)比其相应反式位置(“反反”、“顺反”构象)的RC3-N2长了0.001 1~0.001 3 nm,键角AC2-N1-C1要大1.0° ~1.5°.这是由于芳香性取代基处于反式构象时,由于分子内氢键存在,与脲基处于同一平面,增强了脲基与芳环之间的电子离域,进而增强了脲基与芳环之间键的强度.由于C=S接受氢能力比C=O弱,硫脲受体2缺少分子内氢键存在,因此即使“反反”构象也不能维持平面构型.由于缺少分子内氢键的稳定作用,受体2“反反”构象的能量反而略高于“反顺”构象,且4种构象相应的键长、键角变化不如受体1明显.
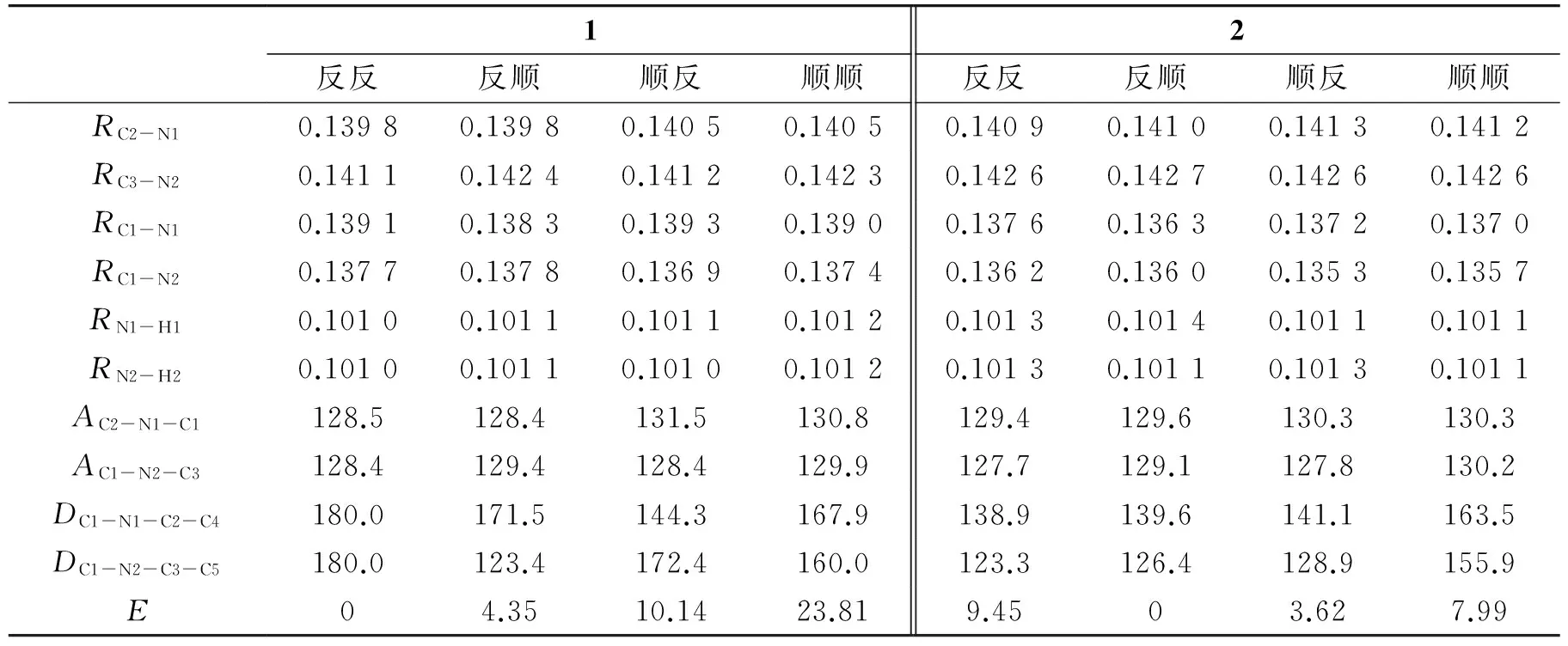
表1 受体1、2四种构象的一些关键几何参数和相对能量(键长:nm,键角和二面角:°,能量:kJ·mol-1)Table 1 Selective geometrical parameters and relative energies of four stable conformers of receptor 1 and 2. (bond length in nm,angle in °,and energies in kJ·mol-1)
受体分子1、2与F-、Cl-阴离子形成的氢键复合物的优化结构见图2,一些关键的几何参数见表2.为了方便与氢键复合物比较,图2中受体2为“反反”构象,而非最稳定“反顺”构象.为了加以区分,在下面的讨论中,我们将以a代表受体1的“反反”构象,而b代表受体2的“反反”构象.尽管受体2具有“反顺”结构的最稳定构象,但其与阴离子作用时,“反反”构象更稳定.这是由于“反反”构象能够与阴离子形成双氢键结构,而“反顺”构象只能形成单一氢键.这意味着,随着阴离子的加入,受体2分子将发生由“反顺”向“反反”构象转变的异构化过程.
从图2和表2中可以看出,在与阴离子(F-、Cl-)形成氢键复合物之后受体a两侧所成二面角仍为180.0°,结构并未发生明显变化;而受体b与F-形成氢键复合物之后,两侧二面角分别为162.0°、137.5°,而在与Cl-形成氢键复合物之后两侧的二面角都为180.0°,较原先的138.9°和123.3°相比,更趋向于形成稳定的平面结构,更有利于形成双氢键结构.对于所形成的氢键来说,在a…F-中,RH1…F和RH2…F的值分别为0.163 3和0.170 3 nm;但在a…Cl-中,左右两侧形成的氢键键长RH1…Cl和RH2…Cl的值分别为0.228 1和0.231 9 nm.同样的对于b…F-,RH1…F和RH2…F的值分别为0.160 5和0.167 0 nm,b…Cl-中RH1…Cl和RH2…Cl的值分别为0.225 1和0.228 5 nm.说明氟离子形成的氢键远强于氯离子的氢键;受体与F-、Cl-形成的都是不对称的双氢键,且异吲哚一侧的氢键相较于苯基一侧更强一些.除此之外,我们还可看出受体b与F-形成的氢键键长比受体a与F-的氢键短了0.002 8~0.003 3 nm,同样受体b与Cl-形成的双氢键的键长比受体a相应的键长要短0.003 0~0.003 4 nm,故可证明受体b与阴离子(F-、Cl-)形成的氢键比受体a形成的氢键要强,这与硫脲基酸性比脲基更强是一致的.
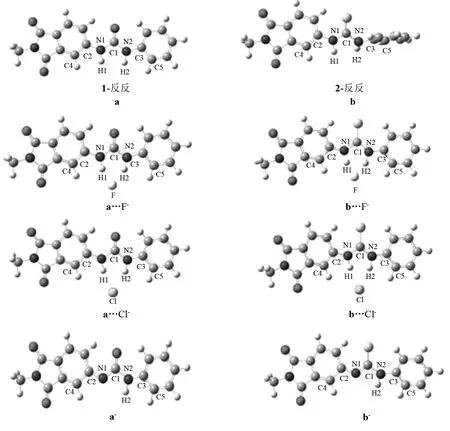
图2 受体1和2与卤素阴离子(F-、Cl-)形成的氢键复合物及其去质子化产物的稳定构象Fig.2 The optimized geometrical structures of the hydrogen bond complexes and the deprotonated products of the receptors 1 and 2
GOMEZ等[12]的研究指出,当F-过量时,脲或硫脲基易发生去质子化过程.计算结果表明,对于受体a和b脱去左边(即异吲哚一侧)的质子所形成的阴离子(a-、b-)的产物要比脱去右边(即苯基一侧)的质子的产物的能量分别低约12.60 kJ·mol-1、9.97 kJ·mol-1.故不论受体a或受体b去质子化过程脱去的都是左侧的质子.表2中列出了的数据表明,对于受体a和受体b,脱去质子后, C2-N1、C3-N2、C1-N1等键的键长均有明显的缩短,而C1-N2则明显的增长.如阴离子a-的RC2-N1、RC3-N2、RC1-N1的键长比对应的受体a分别短0.003 6、0.001 7、0.003 6 nm,而RC1-N2长了0.003 1 nm.
2.2 氢键相互作用能及分子振动特性分析
表3中给出了4种氢键复合物的氢键能(ΔE)和经过CP方法对BSSE校正后的氢键能(ΔECP).从表3中数据我们可以看出对BSSE进行校正之后,氢键能(ΔECP)相较于未校正的ΔE有了明显的增加.受体与不同阴离子所形成的氢键强度(ΔECP)也有很大的差别,如受体a与F-形成的氢键复合物a…F-的ΔECP为-282.89 kJ·mol-1,而与Cl-所形成的氢键复合物a…Cl-的ΔECP则比a…F-的弱87.87 kJ·mol-1.这是由于,与Cl-离子相比,F-半径更小,电负性更大,是更强的氢键接受体.再来对比受体a和受体b与同一种阴离子F-形成的两种氢键复合物a…F-和b…F-的相对大小,结果表明a…F-的ΔECP要比b…F-弱10.41 kJ·mol-1,故受体b与F-形成的双氢键要比相应的受体a更强,对于Cl-也如此.这是因为在DMSO中硫脲的pKa为21.0,而尿素的pKa为26.9,[17]硫脲基的酸性比脲基更强,与F-、Cl-的结合能力更强.
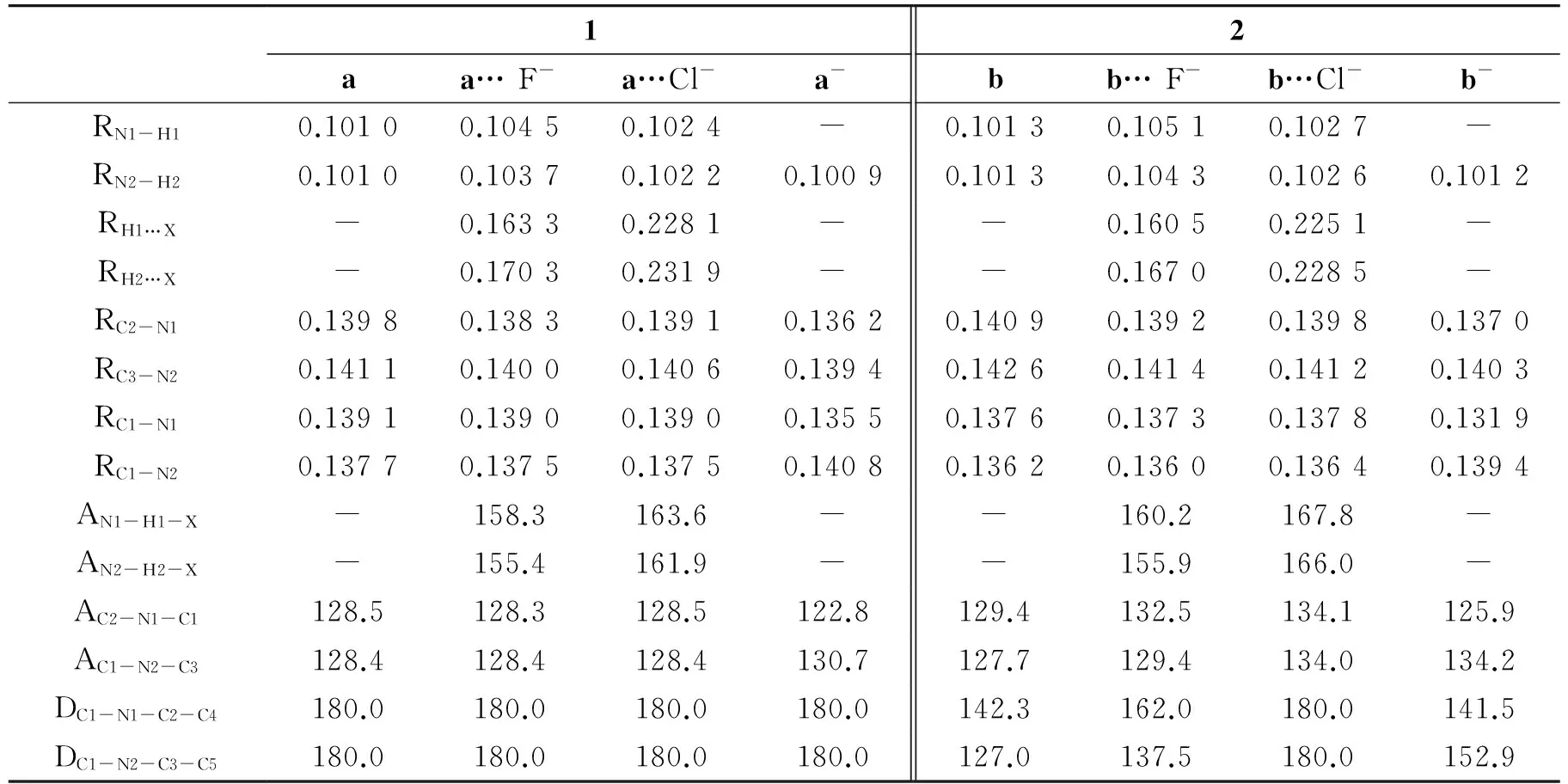
表2 受体1、2与阴离子形成的氢键复合物及其去质子化后体系的部分关键键参数(键长:nm,键角和二面角:°)Table 2 Selected geometrical parameters of the hydrogen complexes and the deprotonated products of the receptors 1 and 2 (bond length in Å and angles in °)
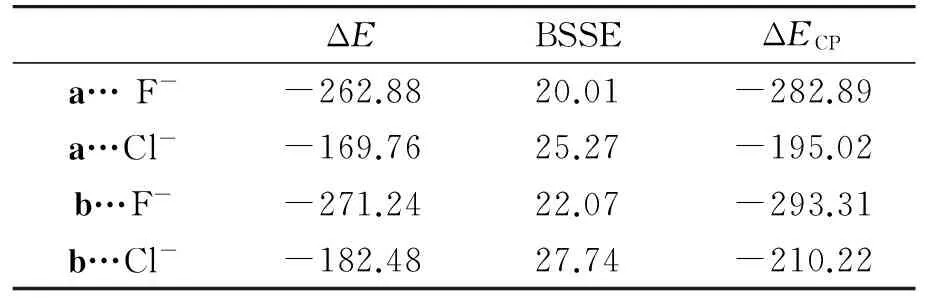
表3 几个氢键复合物的相互作用能ΔE和经CP校正的氢键相互作用能ΔECP(单位:kJ·mol-1)Table 3 Uncorrected and corrected interaction energies with counterpoise procedure of the hydrogen complexes (in kJ·mol-1)
表4中给出了受体a和b及其与阴离子形成氢键复合物后的N-H键的对称和反对称伸缩振动频率(v,cm-1)及其频率移动 (Δv,cm-1).在氢键研究中,氢键给体伸缩振动的红移是氢键的重要特征之一,也是识别氢键存在的主要依据.从表4可以看出,受体a和b在与卤素阴离子形成氢键复合物之后,N-H键的伸缩振动频率vN-H明显发生了红移.如在a…F-和a…Cl-的识别体系中,N-H键的对称伸缩振动频率vN-H红移值分别高达626 和280 cm-1,反对称伸缩振动频率发的红移值则分别487和243 cm-1.F-离子氢键的红移值远大于Cl-离子氢键,这与受体a、b与F-离子形成氢键更强是一致的;而受体b形成的氢键对应的红移值也略大于受体a形成的氢键,这与受体b的氢键强于受体a一致.
表4 受体a、b及其与 F-、Cl-的氢键复合物中N-H键的伸缩振动频率(v:cm-1)和频率移动(Δv:cm-1)
Table 4 Stretch vibrational frequencies (ν) of N-H of the receptors a and b,and their complexes and the frequency
shifts (Δν) caused by the hydrogen bonds (in cm-1)
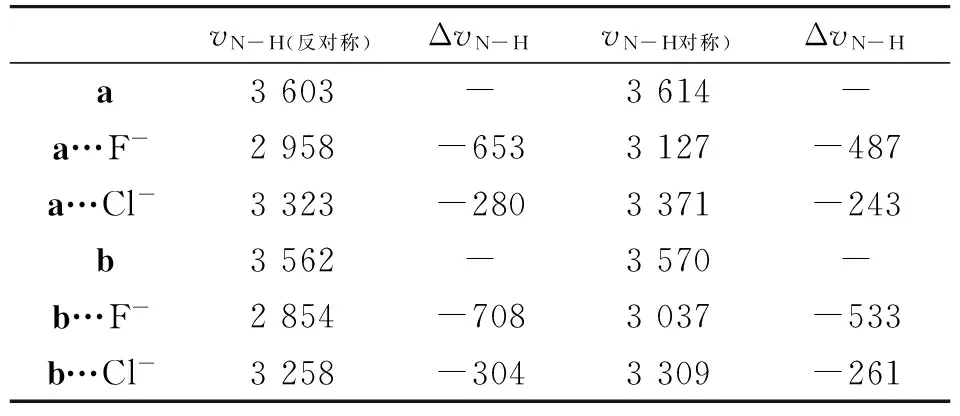
vN-H(反对称)ΔvN-HvN-H对称)ΔvN-Ha3603-3614-a…F-2958-6533127-487a…Cl-3323-2803371-243b3562-3570-b…F-2854-7083037-533b…Cl-3258-3043309-261
2.3 UV-Vis光谱分析
分别采用TD-B3LYP和TD-CAM-B3LYP的方法计算了受体a和受体b及其与阴离子(F-、Cl-)的氢键复合物的UV-Vis光谱.结果表明长程校正对体系的UV-Vis有很大的影响,故之后的数据均采用CAM-B3LYP方法计算的数据.表5中列出了受体a、b与阴离子(F-、Cl-)形成氢键复合物及其去质子化后所形成体系主要的电子跃迁.由表中数据可知,受体a、b及其氢键复合物的第一吸收峰主要来源于电子从最高占据轨道(HOMO)到最低空轨道(LUMO)的跃迁,但受体a、b在与阴离子形成氢键复合物及其去质子化之后都使第一吸收峰发生了明显的红移.
对于受体a,在未加入阴离子前第一吸收峰为320 nm,电子跃迁也并不复杂,主要为H → L的跃迁.加入F-后所形成的氢键复合物a…F-的第一吸收峰为340 nm,红移值为20 nm;加入Cl-之后所形成氢键复合物a…Cl-的第一吸收峰为329 nm,红移值为9 nm.对于受体b,通过之前的计算我们已经知道对于受体2来说在未加入阴离子之前在溶液中的存在形式为“反顺”构型,此结构的第一吸收波长为323 nm,而“反反”(即b)结构的第一吸收峰为331 nm,电子跃迁类型为H → L的跃迁.加入Cl-之后,复合物的H→L跃迁的吸收峰位于331 nm,这一数值相对于“反反”构象几乎没有变化,但相对于“反顺”构象红移了8 nm.我们相信这一红移现象是由于受体分子构象变化引起,而与氢键无关.加入F-后,复合物的电子跃迁相对于最稳定的“反顺”构象红移了19 nm;而相对于“反反”构象也红移了10 nm,这部分红移由氢键引起.通过以上分析,受体a和受体b与F-形成氢键复合物能够产生较大的电子光谱红移,但与Cl-形成氢键的红移值比较小,特别是b…Cl-复合物中的氢键几乎对电子光谱没有任何改变.这是由于受体a和受体b与F-所形成的双氢键的强度要远大于与Cl-所形成的双氢键的强度.
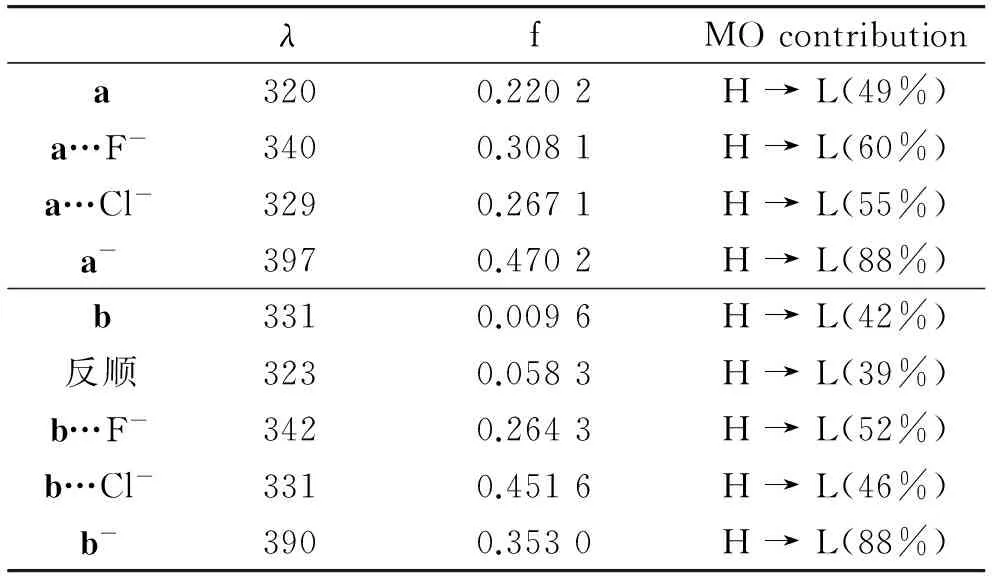
表5 受体a和b及其与阴离子复合物的吸收波长(nm)、振子强度、HOMO→LUMO跃迁的贡献率 (H:HOMO,L:LUMO)Table 5 Absorption wavelength(nm),the oscillator strength, and the contribution of HOMO→LUMO transition of the receptors a and b and their complexes (H:HO-MO,L:LUMO)
当加入过量的F-之后,受体a和受体b均发生了去质子化过程,对与受体a去质子化后所形成的阴离子a-的第一吸收峰为398 nm,相对于和F-形成的氢键复合物a…F-又发生了明显的红移,红移值高达58 nm.对于受体b,当加入过量的F-之后,所形成的阴离子b-的第一吸收峰为390 nm,相较于b…F-发生了很大程度的红移,红移值高达49 nm.GOMEZ等[12]通过做实验得出受体b在DMSO中的第一吸收波长为330 nm,与我们的计算值323 nm相近,而当加入F-过量导致去质子化之后的第一吸收波长为410 nm,与计算所得的390 nm也很接近,理论值与实验值能很好地吻合.
2.4 前线轨道分析
图3显示了受体a、受体b及其氢键复合物、去质子化产物的分子轨道.通过图3进行分析,我们得知4种体系的分子轨道形状基本相同,其中HOMO轨道分布于整个分子,而LUMO轨道定域在异吲哚环的部分.从图3可以看出,受体a的第一激发态对应着π-π*电子跃迁,并伴随着一定的分子内电荷转移,即部分电荷从右边的苯环转移到左边的吲哚环.受体b与a类似,HOMO到LUMO的跃迁属于π-π*电子跃迁,伴随着一定的电荷转移现象,这些电荷转移过程必然导致激发态体系极性及溶剂化效应的改变.此外,受体与阴离子间的氢键对受体分子的HOMO与LUMO能隙产生了较为明显的影响.受体a和b的HOMO与LUMO能隙分别为3.70和3.59 eV.与F-、Cl-离子间的氢键导致a的能隙分别降低了0.27和0.14 eV,而去质子化过程使a的能隙降低了0.64 eV.类似地,与F-离子的氢键使受体b的能隙降低了0.17 eV,但Cl-离子几乎没有改变受体b的能隙.这些结果与受体a、b及其与F-、Cl-离子氢键复合物的吸收光谱红移规律是一致的,可见氢键和去质子化导致的受体分子HOMO与LUMO能隙减小是受体a、b吸收光谱红移的主要原因.
3 结论
采用密度泛函理论对基于脲及硫脲衍生物的两个阴离子受体分子进行了系统的计算研究得出以下结论:
1) 由于分子内氢键Cα-H…O=C的存在,基于脲衍生物的受体1倾向于形成“反反”构象的平面构型;而硫脲基作为氢键受体能力明显小于脲基,因而基于硫脲衍生物的受体2没有分子内氢键形成,其最稳构象为扭曲的“反顺”构象.
2) 受体1、2均能与卤素阴离子(F-、Cl-)形成稳定的双氢键结构,但是与F-离子的氢键强度远大于Cl-离子;此外,硫脲基酸性略强于脲基,因此受体2形成的氢键强于受体1.

图3 受体a、b,及其与阴离子形成氢键复合物,及其去质子化后形成阴离子的前线分子轨道Fig.3 Frontier molecular orbitals of the receptors a and b,their hydrogen bond complexes,and their deprotonation products
3) 受体1、2与F-离子间的强氢键导致受体分子吸收光谱的明显红移,而与Cl-离子间的氢键较弱,相应的光谱红移程度较小.此外,随着过量F-离子的加入,受体1、2发生去质子化过程,导致吸收光谱的进一步红移.
[1] SESSLER J L,GALE P,CHO W S.Anion receptor chemistry [M].Cambridge:The Royal Society of Chemistry,2006:1-26.
[2] GALE P A,BUSSCHAERT N,HAYNES C J E,et al.Anion receptor chemistry:highlights from 2011 and 2012 [J].Chem Soc Rev,2014,43(1):205-241.
[3] 李勇军,刘辉彪,李玉良.基于氢键的阴离子识别主体分子的研究进展[J].无机化学学报,2015,31(9):1687-1704.
LI Y J,LIU H B,LI Y L.Recent development of anion receptors based on hydrogen bonding [J].Chinese Journal of Inorganic Chemistry,2015,31(9):1687-1704.
[4] 李玉玲,刘克成,孙红先,等.酰胺类阴离子受体的研究进展[J].南阳师范学院学报,2012,11(3):48-53.
LI Y L,LIU K C,SUN H X,et al.Research on the amide anion receptors [J].Journal of Nanyang Normal University,2012,11(3):48-53.
[5] LIU C Y,CHEN T H,MISRA T K.A macrocyclic polyamine as an anion receptor in the capillary electrochromatographic separation of carbohydrates [J].Journal of Chromatography A,2007,1154(1/2):407-415.
[6] 魏梅莹,李少光,贾传东,等.脲类受体对阴离子的结合、识别和分离[J].高等学校化学学报,2011,32(9):1939-1949.
WEI M Y,LI S G,JIA C D,et al.Anion binding,recognition and separation by urea-based receptors [J].Chemical Journal Chinese Universities,2011,32(9):1939-1949.
[7] 吴芳英,温珍昌,江云宝.硫脲类阴离子受体的研究进展[J].化学进展,2004,16(5):776-784.
WU F Y,WEN Z C,JIANG Y B.Thiourea-based receptors for anion recognition and sensing [J].Progress in Chemistry,2004,16(5):776-784.
[8] 巫文静,鲁润华,高海翔,等.胍基化合物在分子识别中的应用[J].合成化学,2005,13(6):529-535.
WU W J,LU R H,GAO H X,et al.Application of guanidines in molecular recognition [J].Chinese Journal of Synthetic Chemistry,2005,13(6):529-535.
[9] 魏太保,王军,郭潇迪,等.含酚羟基Schiff碱化合物的阴离子识别研究[J].化学研究与应用,2008,20(7):858-861.
WEI T B,WANG J,GUO X D,et al.Anion recognition of Schiff base bearing phenolic hydroxyl groups [J].Chemical Research and Application,2008,20(7):858-861.
[10] GHOSH A,JOSE D A,DAS A,et al.A density functional study towards substituent effects on anion sensing with urea receptors [J].Journal of Molecular Modeling,2010,16(9):1441-1448.
[11] LI G Y,CHU T.TD-DFT study on fluoride-sensing mechanism of 2-(2[prime or minute]-phenylureaphenyl) benzoxazole:the way to inhibit the ESIPT process [J].Physical Chemistry Chemical Physics,2011,13(46):20766-20771.
[12] GOMEZ D E,FABBRIZZI L,LICCHELLI M,et al.Urea vs.thiourea in anion recognition [J].Organic & Biomolecular Chemistry,2005,3(8):1495-1500.
[13] YANAI T,TEW D P,HANDY N C.A new hybrid exchange-correlation functional using the coulomb-attenuating method (CAM-B3LYP) [J].Chemical Physics Letters,2004,393(1/3):51-57.
[14] ROSTOV I V,AMOS R D,KOBAYASHI R,et al.Stu-dies of the ground and excited-state surfaces of the retinal chromophore using CAM-B3LYP [J].The Journal of Physical Chemistry B,2010,114(16):5547-5555.
[15] JACQUEMIN D,PERPETE E A,SCUSERIA G E,et al.TD-DFT performance for the visible absorption spectra of organic dyes:conventional versus long-range hybrids [J].Journal of Chemical Theory and Computation,2008,4(1):123-135.
[16] FRISCH M J,NAKATSUJI M,CARICATO X,et al.Gaussian 09 [CP].Revision E.01,Wallingford CT:Gaussian,Inc,2013.
[17] BORDWELL F G.Equilibrium acidities in dimethyl sulfoxide solution [J].Accounts Chemical Research,1988,21(12):456-463.
[责任编辑:吴文鹏]
DFT study on anion receptors based on urea and thiourea derivatives
GENG Weihong,WANG Yaru,LI Xia,ZHANG Ruihong,WANG Bingqiang*
(SchoolofChemistryandMaterialScience,ShanxiNormalUniversity,Linfen041000,Shanxi,China)
Anion recognition process of two receptors based on the urea and thiourea derivatives was studied using density functional theory (DFT).The urea derivative (receptor 1) is a “trans,trans” conformer bearing two Cα-H…O=C intramolecular hydrogen bonds.Otherwise,for the thiourea deri-vative (receptor 2),the “trans,cis” conformer is the most stable due to absence of intramolecular hydrogen bonds.The receptors 1 and 2 both form double hydrogen bonds with F-and Cl-,and meanwhile the receptor 2 would change its conformer from “trans,cis” to “trans,trans”.Moreover,the hydrogen bonds of F-anion are significantly stronger than those of Cl-anion,and the hydrogen bonds of thiourea donor group are stronger than those of urea group.Finally,a formation of hydrogen bonds and the de-protonation process with F-cause a large red-shift of absorption spectra,however the interaction with Cl-has a small effect on the spectra.
urea; thiourea; anion receptor; density functional theory
2016-09-29.
山西师范大学大学生创新创业训练项目(SD2014CXXM-45).
耿玮宏(1994-),女,研究方向为量子化学计算.*
,E-mail:wangbq2007@163.com.
O641
A
1008-1011(2017)01-0019-08
猜你喜欢
化工管理(2021年7期)2021-05-13 00:45:38
农药科学与管理(2019年8期)2019-11-23 08:04:44
红外技术(2017年1期)2017-03-27 09:02:21
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
China International Studies(2016年3期)2016-07-14 03:00:06
质谱学报(2015年5期)2015-03-01 03:18:25
应用化工(2014年7期)2014-08-09 09:20:23
无机化学学报(2014年5期)2014-02-28 17:31:40
科技资讯(2013年7期)2013-04-29 00:44:03
