
一种新型开环烯二炔的设计、合成及细胞毒性研究
2017-03-08 03:45李保君胡爱国
华东理工大学学报(自然科学版) 2017年1期
黄 帅, 李 竞, 李保君, 胡爱国
(华东理工大学材料科学与工程学院,上海市先进聚合物材料重点实验室,上海 200237)
一种新型开环烯二炔的设计、合成及细胞毒性研究
黄 帅, 李 竞, 李保君, 胡爱国
(华东理工大学材料科学与工程学院,上海市先进聚合物材料重点实验室,上海 200237)
通过Sonogashira反应合成了一种以马来酰亚胺为主体结构,炔基带有两个对称烷基链的烯二炔化合物。该烯二炔在适度加热的情况下能够发生Bergman环化反应,产生芳基双自由基。通过红外光谱分析确认反应后碳碳叁键信号峰消失;利用自由基捕捉技术获得了单N-叔丁基-α-苯基硝酮(PBN)加成产物和双PBN加成产物,并通过电子顺磁共振波谱检测到了这两种PBN加合物的自由基信号,说明烯二炔发生了Bergman环化反应。MTT法细胞活性实验证实该小分子烯二炔对肿瘤细胞的半抑制浓度在18 μmol/L左右,显示出烯二炔对肿瘤细胞具有良好的抗增殖能力。这为以后设计合成高活性的开环型烯二炔抗生素提供了新的思路。
烯二炔; Bergman环化; 抗肿瘤抗生素; 细胞毒性
多年来,世界各国都在关注如何根治癌症。癌症是引起发达国家人口死亡的主要原因,在发展中国家癌症的发病率也在不断增加[1]。治疗癌症的方式多种多样,其中化学治疗是十分重要的手段之一。化学治疗癌症的常用药物是抗生素。药用抗生素绝大部分含有天然的活性成分,可以影响癌细胞的生长行为。1965年,Ishida等[2]从链霉菌(Streptomycescarzinostaticus)的发酵液中分离出新制癌菌素(Neocarzinostatin,NCS)。随后人们陆续发现了Neocarcinostatin Chromophore[3]、Esperamicins[4]、Calicheamicins[5]、Dynemicins[6]、Kedarcidin Chromophore[7]、Lidamycin[8]等20多种天然烯二炔类抗肿瘤抗生素。这些天然抗生素都有一个相似的主体化学结构——烯二炔,该基团可以被靶向传输到DNA的小沟内,引发Bergman或者Myers-Saito环芳香化反应,产生高度活性的苯基双自由基。这些自由基可以进一步夺取磷酸骨架上的氢原子,导致DNA的单链或者双链裂解,引起细胞的凋亡。天然烯二炔抗生素的抗肿瘤活性甚至比临床上最为有效的阿霉素高出5~8 000倍[9]。例如:体外试验表明C-1027对于KB肿瘤细胞有着极其突出的细胞毒性,其半抑制浓度(IC50)甚至低于0.1 ng/mL[10]。体内试验表明该抗生素对于荷瘤小鼠也展示出了高效的抗肿瘤活性[11]。虽然天然烯二炔抗生素对多种肿瘤细胞都表现出强烈的毒性,但是其缺乏细胞选择性的弊端使得它们在杀死肿瘤细胞的同时也对正常细胞显示出细胞毒性[9,12-13],并且伴随有一定的副作用,如白细胞减少[14]、心肌病等[15]。
天然烯二炔抗生素的独特抗肿瘤机制和它们显而易见的弊端为有机合成化学家们尝试人工合成烯二炔提供了机遇和挑战。过去几十年来,人们合成出了各种类型的烯二炔抗生素,希望在合适的引发条件下能够提高Bergman环化反应的活性,并且能够降低烯二炔对于正常细胞的毒性。其中研究较为广泛的是pH引发机制和光照引发机制[16]。
Basak等[17]报道了可以根据肿瘤细胞中的酸性环境设计出pH响应的烯二炔,合成的几种烯二炔在碱性环境下对所测细胞没有表现出明显的毒性优势。当引发条件置换为弱碱环境,目标烯二炔开始发生Bergman环化反应,产生细胞毒性,并且对肿瘤细胞的半抑制浓度达到了临床上的要求。但是,这些烯二炔在保持高毒性时,不能区分正常细胞和肿瘤细胞。Jones等[18]设计合成了一种可以定向攻击肿瘤细胞特征区域的烯二炔,这些区域包括像核酸靶体突起的地方,在与这些核酸突起相互作用的过程中,烯二炔分子发生了构型转变,使得烯二炔的炔基末端处在一个临界的碳原子之间的距离(简称cd距离),进而引发Bergman环化反应。Sharma等[19]设计合成了多种十三元的环状烯二炔抗生素,展现出了对既选细胞良好的抗增殖能力。在靶向区域,作者使用了外部光源来触发烯二炔的反应活性。该种设计方案成功的前提条件是在特定波长的光源作用烯二炔之前,烯二炔小分子能够稳定存在。同时,外部的光源照射不能对细胞产生额外的毒性。这种方法证明了光照引发可以选择性地作用特定部位,但是受限于光源的穿透组织的能力,光源到达不了所有的肿瘤部位。Polukhtine等[20]通过调整取代基的电子性能合成出了一种新型的烯二炔,提高了环化产物的产率。他们通过不同模式的激发能量转移,旨在减轻烯二炔毒性,并且在时间和空间上控制反应活性。在测试肿瘤细胞毒性时,羰基结构的烯二炔没有破坏DNA,光照以后生成的羟基结构的烯二炔对DNA产生了稳健的单链和双链裂解能力。
近期,我们研究组[21]设计并合成出了一种含有马来酰亚胺以及三乙氧基官能团的开环烯二炔。这种烯二炔的三乙氧基官能团可以在肿瘤细胞中的酸性部分水解,形成双羰基结构。羰基强烈的吸电子作用使其能够在常温下引发Bergman环化反应。之后,又将马来酰亚胺部分的封端苯基替换成活性较高的呋喃环[22]。令人惊喜的是,这种烯二炔不仅保留了Bergman反应的活性,而且表现出了较好的细胞选择性。在前期研究工作的基础上,本文选择以马来酰亚胺为主体结构,通过寻找合适的炔基取代基,统筹考虑cd距离、空间位阻、电子效应等因素,合成了具有如下特点的开环烯二炔:开环烯二炔化合物能够在相对较低的温度范围内发生Bergman环化反应,引入马来酰亚胺基团可明显地降低分子发生Bergman环化反应的能垒[23];合成的烯二炔具有对称的长链烷基,可以降低烯二炔分子的极性表面积,有助于分子渗透入亲脂性的细胞膜。
1 实验部分
1.1 主要仪器与试剂
1.1.1 主要原料 1-己炔,纯度98%,安耐吉化学有限公司;2,3-二氯马来酸酐,纯度99.9%,安耐吉化学有限公司;N,N-二异丙基乙胺,纯度98%,安耐吉化学有限公司;碘化亚铜,纯度99%,国药集团化学试剂有限公司;苄胺,纯度99%,阿拉丁化学试剂有限公司;N-叔丁基-α-苯基硝酮(PBN),纯度99.9%,阿拉丁化学试剂有限公司;乙腈、乙酸,分析纯,上海凌风化学试剂有限公司;硅胶(50~75 μm)、无水硫酸镁,分析纯,国药集团化学试剂有限公司;氘代试剂,分析纯,百灵威科技有限公司;石油醚、乙酸乙酯、正己烷、二氯甲烷,分析纯,苏州晶协化学有限公司;甲苯、四氢呋喃(THF)在使用前经干燥处理,常压蒸馏后方可使用。
1.1.2 主要仪器 核磁共振氢谱和碳谱(1H-NMR,13C-NMR):采用美国Bruker公司的核磁共振仪(BRUKER BIOSPIN AG,Magnet System 400 MHz/500 MHz),测试所用溶剂为氘代氯仿(CDCl3)。
色谱-质谱联用:采用英国Micromass LCTTM型液相色谱-飞行时间质谱联用仪,配以Waters 600液相色谱仪进样系统,离子化方式为电喷雾电离(ESI)。
基质辅助激光解析-飞行时间质谱:采用美国AB Sciex公司生产的基质辅助激光解析-飞行时间质谱仪(4 800 plus)。
差示扫描量热分析(DSC):采用德国微分扫描量热分析仪Pyris钻石热分析工作站进行检测,装配有822e DSC模块模型,氮气气氛下检测温度为室温至250 ℃,升温速率10 ℃/min。
傅里叶变换红外光谱(FT-IR):采用美国Thermo Nicolet公司生产的Nicolet 5700型傅里叶变换红外光谱仪,通过KBr压片法以及涂膜法制样,测定透射光谱。
紫外可见光谱(UV-Vis):采用Shimadzu UV-2550紫外-可见分光计。
电子顺磁共振波谱(EPR):将待测物溶解在三氯甲烷(10 mmol/L)中进行测试。使用EMX-8 /2.7C的EPR光谱仪(Bruker公司,德国),选择在X波段测量。光谱仪设置为:扫描宽度0.01 T,时间常数163.84 ms,转换时间40.96 ms,分辨率1 024点,调制频率100.00 kHz,调制振幅10-4T,微波频率9.877 GHz,微波功率6.358 mW。
1.2 烯二炔抗生素的合成
烯二炔抗生素的合成步骤如图1所示。
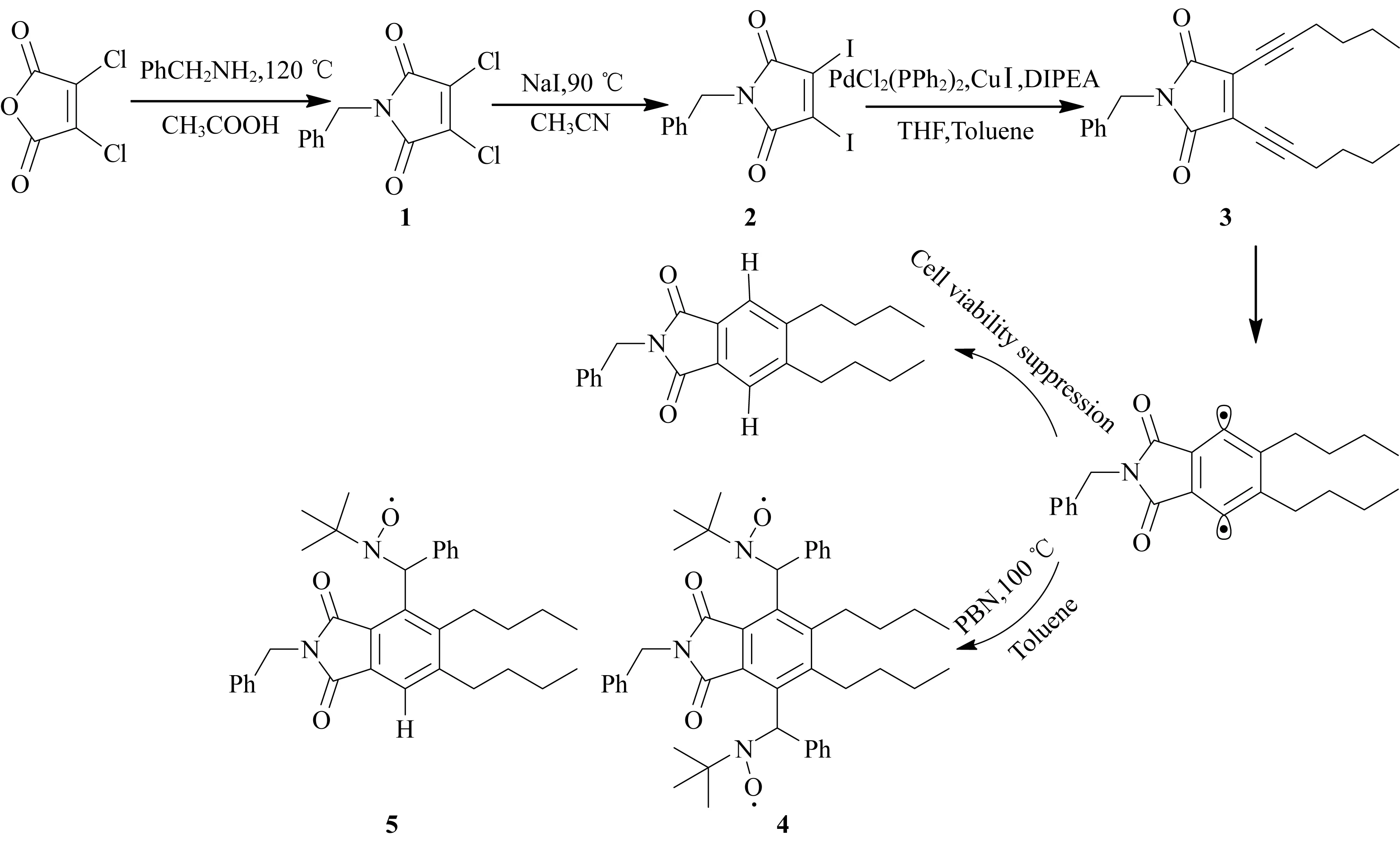
图1 烯二炔抗生素的合成路线和Bergman环化反应
化合物1[24]:称取2,3-二氯马来酸酐(6.01 g,36 mmol)和40 mL乙酸,加入到100 mL圆底烧瓶中,室温搅拌至完全溶解。将苄胺(4 mL,36.6 mmol)缓慢滴加到圆底烧瓶中,立刻有大量白雾产生,室温搅拌至白雾消失。慢慢添加苄胺,待全部滴加完毕后,将整个反应体系转移至120 ℃油浴搅拌器中回流反应22 h。反应结束后,逐渐降低油浴锅温度使得体系冷却至室温,真空挥发除去乙酸,用乙酸乙酯作为良溶剂溶解整个粗产物,分液萃取后得到的固体经柱层析硅胶色谱柱分离,以正己烷和乙酸乙酯(正己烷与乙酸乙酯的体积比15∶1)为淋洗剂洗脱,最终分离得到产物1(6.27 g,质量产率68.1%)。1H-NMR(CDCl3,400 MHz,δ):7.36~7.28(m,5H),4.69(s,2H)。
化合物2[24]:称取二氯马来酰亚胺1(2.29 g,8.95 mmol)和40 mL乙腈至100 mL圆底烧瓶中,添加碘化钠(5.05 g,35.8 mmol)后,将整个反应体系置于90 ℃油浴搅拌器中,避光回流反应5 h。待反应结束后,逐渐降低油浴锅温度使得体系冷却至室温,快速将溶液倾入大量去离子水中,在玻璃壁上迅速有黄色絮状沉淀析出。快速搅拌24 h后,减压过滤,至水分基本风干后,得到的滤饼在真空烘箱中干燥5 h,得到橙黄色粉末固体产物2(2.69 g,质量产率68.4%)。1H-NMR(CDCl3,400 MHz,δ):7.31~7.22(m,5H),4.70(s,2H)。
化合物 3[25]:称取化合物2(632 mg,1.44 mmol),碘化亚铜(28 mg,0.144 mmol),二(三苯基磷)二氯化钯(50 mg,0.072 mmol),N,N-二异丙基乙胺(DIPEA,622 μL,3.624 mmol)依次加入到50 mL的Schlenk瓶中,真空线下抽真空-置换氮气数次,在氮气氛围下加入用氢化钙处理过的THF(3 mL)和用钠砂处理后的甲苯(10 mL),搅拌溶解,最后加入己-1-炔(441 μL,3.624 mmol)。氮气保护下在45 ℃搅拌反应,用薄层层析法(TLC)跟踪至完全反应。反应停止后,将上层褐色液体直接通过柱层析硅胶柱色谱进行分离,用石油醚和乙酸乙酯(体积比30∶1)为淋洗剂洗脱,得到浅黄色黏稠液体304 mg,质量产率为61%。1H-NMR(CDCl3,400 MHz,δ):7.36~7.26(m,5H),4.67(s,2H),2.55(t,4H),1.61(m,4H),1.49(m,4H),0.94(t,6H);13C-NMR(CDCl3,125 MHz,δ):167.3,135.8,128.6,128.5,127.9,112.1,71.7,42.2,30.1,21.9,20.1,13.5;HRMS(ESI):m/zcalcd.for C13H26NO2[M+H]+:348.196 4,found:348.196 8。
PBN加合物(化合物4和5)[26]:称取化合物3(156 mg,0.449 mmol)溶解在40 mL甲苯溶剂中,加入到50 mL封口瓶中,充分搅拌溶解以后,立即将PBN(199 mg,1.123 mmol)加入到封口瓶中,真空线下抽真空-置换氮气数次。氮气气氛下油浴100 ℃搅拌反应24 h。停止反应后,真空挥发除去溶剂,用饱和氯化钠溶液和乙酸乙酯萃取数次后,取有机层溶液,经无水硫酸镁干燥、过滤、旋蒸除去溶剂。通过柱层析硅胶色谱柱进行分离,用石油醚和乙酸乙酯(体积比25∶1)为淋洗剂洗脱,收集得到橙红色黏稠液体56.8 mg,经过EPR证实该产物是PBN的加合物(化合物4和5的混合物)。在高分辨质谱中出现化合物5的峰,m/zcalcd.for C34H41N2O3[M]+:525.311 7;found:525.290 5。
1.3 细胞活性实验
贴壁细胞MTT实验:在细胞实验开始之前,本实验所用细胞株都必须预先解冻、生长、分裂,以获得足够数目的细胞用于细胞实验。细胞株经充分生长后接种到96孔板中,接种密度为3 000。再将细胞放置在培养箱里贴壁培养,培养箱里以CO2气体氛围填充。与此同时,配制浓度为100 mmol/L的烯二炔合化物3的丙酮溶液,经0.22 μm微孔滤膜过滤以后,逐步稀释到100 μmol/L,再次稀释到0.3、0.6、1.2、2.3、4.7、9.4、18.8、37.6 μmol/L,另外在作为参考样的丙酮中加入培养液稀释到丙酮体积分数为0.1%。经过1 d的细胞培养,除去96孔板中的培养液介质,取代以200 μL各种浓度的烯二炔化合物3的丙酮溶液。空白组实验则加入200 μL体积分数为0.1%的丙酮溶液,转移孔板至CO2培养箱里培养2 d。培养过程结束后,向每个实验孔板中添加20 μL左右的 MTT试剂,另外单独培养4 h。取出培养细胞的孔板,除去每个孔板中的培养液,待培养液全部除去后,适时向板孔中添加200 μL左右的二甲基亚砜溶液。以上操作完成以后,将整个孔板置于室温环境下,平铺在超净台面上,等待约10 min。再将孔板置于酶标仪中,匀速摇荡整个体系30 s,直至甲臜结晶物在溶剂中逐渐消失。调节仪器至波长490 nm处,对准每个孔板计算出它们的吸光度值大小。将细胞的吸光度值对比空白组的吸光度值并以百分比来表示,得到最终结果。
2 结果与讨论
2.1 化合物的合成
由Nicolaou提出的烯二炔化合物的间距理论认为,烯二炔化合物能否自发发生Bergman环化反应取决于两个叁键末端(1,6位)的cd距离[27]。Schreiner等[28]指出影响热致Bergman环化反应的因素分别是cd距离、环张力能、电子效应、位阻效应等。开环烯二炔在结构上比环状烯二炔(天然烯二炔均为环状烯二炔)简单很多,也比较容易合成,但是开环烯二炔炔臂间的cd距离大大超出了Nicolaou提出的临界范围,使得开环烯二炔发生热致Bergman环化反应的触发温度比环状烯二炔高得多(一般在200 ℃以上)。因此,合成一种结构简单、作用高效的开环烯二炔抗生素面临着不小的挑战。
本文在烯二炔化合物3的烯烃部位引入马来酰亚胺结构,增强了整个分子共轭区域的吸电子效应,大大降低了热致Bergman环化反应的引发温度。图1显示了整个烯二炔分子的合成路线,通过优化反应条件,确定以二(三苯基磷)二氯化钯为催化剂,N,N-二异丙基乙胺为碱的反应体系获得了较高的产率。在烯二炔3的核磁共振氢谱中,化学位移在7.20~7.35处的信号峰为苯环质子特征峰,化学位移4.7处为与苯环相连的亚甲基的信号,化学位移0.9~2.5左右处的4组峰是典型的烷基质子信号峰。核磁共振碳谱中化学位移在70和110处左右的峰是典型的碳碳叁键信号峰。高分辨质谱中出现了一组峰(质荷比348.196 8),与理论上的C13H26NO2[M+H]+(质荷比348.196 4)接近。以上测试都说明成功合成了烯二炔3。
用差示扫描量热法(DSC)测试证实烯二炔3发生Bergman环化反应的起始温度约为70 ℃左右,相比于之前报道的绝大多数开环烯二炔要低许多。说明马来酰亚胺基团的引入大大降低了其发生热致Bergman环化反应的位垒。由此,我们尝试将烯二炔3在不同极性的溶剂中进行热致Bergman环化反应。在封管中,分别选用了四氯化碳、氯仿、乙腈和二甲基亚砜(DMSO)等溶剂将烯二炔3加热到60 ℃,利用TLC点板跟踪反应。反应24 h以后,发现在极性相对较小的四氯化碳、氯仿溶剂中,原料烯二炔没有发生反应;而在乙腈中,原料烯二炔发生了部分转化;在极性比较大的二甲基亚砜溶剂中,反应6 h后,原料烯二炔就已经反应完全。我们认为烯二炔3在非极性溶剂中能够稳定存在,而当其转移到极性溶剂中,就会加速发生Bergman环化反应。图2示出了烯二炔3及其在极性溶剂二甲基亚砜中加热到60 ℃、反应24 h后所得产物的核磁共振氢谱。对比发现化学位移在7.36~7.26区域的苯环上的氢,化学位移4.67处的苄胺亚甲基的氢,2.55~0.94区域的烷基氢的峰形都展宽了,说明烯二炔发生了Bergman环化反应,苯基双自由基单体之间相互偶联形成结构刚性的聚合物。烯二炔3针对不同极性溶剂表现出的反应活性的差异无法用一般的Bergman环化反应机理来解释[29]。最近,Haberhaueret等[30]提出了烯二炔之间的二聚反应,并且给出了其可能的反应方式。这种反应方式的位垒要远远低于Bergman环化反应,并且每个烯二炔分子只有一个碳碳叁键参与反应,使得最后的聚合物产物中存在丰富的炔基基团。傅里叶红外光谱(图3)显示烯二炔3在2 210 cm-1处有明显C≡C的伸缩振动峰,而聚合物在2 210 cm-1处C≡C峰已经消失,表明并没有发生烯二炔之间的二聚反应。图4示出了将烯二炔3和对应的聚合物溶于甲醇后测得的紫外可见光光谱。实线标记的烯二炔3的光谱表明烯二炔在265 nm 和368 nm处有两组特征的吸收峰,吻合芳香环π-π*过渡态和烯二炔部分的π-π*过渡态。虚线标记的Bergman环化反应之后的聚合物的光谱,呈现出一个拖尾到430 nm位置的宽峰,说明反应前后分子的共轭体系发生了显著的变化。
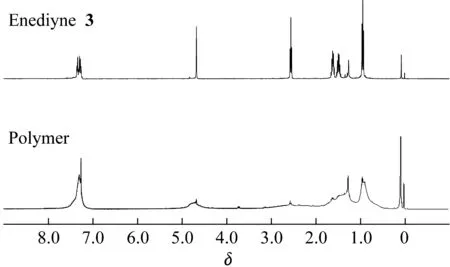
图2 烯二炔3和对应聚合物的核磁共振氢谱

图3 烯二炔3和对应聚合物的红外光谱

图4 烯二炔3和对应聚合物的紫外-可见光谱
为了进一步证实烯二炔3发生了Bergman反应,我们进行了电子顺磁共振波谱(EPR)的测试。热致Bergman环化反应的本质是热致引发条件满足时,会产生高活性的苯基双自由基。自旋捕捉剂可以与寿命短暂的自由基反应,生成相对稳定的自由基加合物[31],再利用EPR检测技术获得相应的加合物图谱,分析出精细的偶合参数,可以判断和识别出自由基的类型,进一步推断出前体的化学结构性质。我们用PBN作为自旋捕捉剂,追踪Bergman反应的中间体的碳自由基[26],由于新生成的氮氧自由基结构比较稳定,可以进一步表征和研究。EPR研究表明(图5),混合物中至少存在两种自由基类型。图5所示的三重精细偶合参数为1.45 mT的峰,和1,4-苯基双自由基的单加成PBN产物非常吻合[26,31-33]。说明了体系中存在单PBN加成的产物。与此同时,在高分辨质谱中也看到了单PBN加成产物的分子离子峰525.290 5。另外,在EPR谱图还可以看到一组微弱的小峰,它们的精细偶合参数为0.56 mT,对应于1,4-苯基双自由基的双PBN加成产物[21,34]。该实验证实烯二炔3发生Bergman环化反应后的自由基可以继续与其他分子反应,为烯二炔3用于生物活性研究提供了支持。

图5 PBN自由基加合物在氯仿中的EPR谱线
2.2 细胞毒性实验
选取了肺腺癌细胞(A549)和正常肝细胞(L-02),运用MTT比色法来评估烯二炔的细胞毒性,并选用丙酮作为有机溶剂载体,传递烯二炔至细胞中。空白实验组表明,采用体积分数为0.1%的丙酮溶液培养细胞以后,肿瘤细胞和正常细胞仍然保持较高的活性。因此,烯二炔3的丙酮溶液如果对细胞产生任何的抑制作用,其活性是来自烯二炔3,而非丙酮。我们尝试将烯二炔3溶解在丙酮溶剂中,制备出新鲜的烯二炔溶液用于细胞毒性的研究。选取不同浓度的烯二炔3对肿瘤细胞A549、正常细胞L-02共同进行培养,图6比较了不同浓度的烯二炔3对肿瘤细胞和正常细胞的抗增殖能力。MTT比色法评价结果显示,抗肿瘤烯二炔3对肿瘤细胞和正常细胞都表现出明显的细胞毒性。当烯二炔浓度在4.7 μmol/L以上,细胞存活率呈现出药物浓度的依赖性。烯二炔3对实验中使用的肿瘤细胞A549的半抑制浓度(IC50)为15~20 μmol/L,凸显了烯二炔3对肿瘤细胞较高的抗增殖能力,说明可以开发烯二炔3用于临床作为抗肿瘤抗生素的潜能。但是,烯二炔3并没有显示出细胞选择性,对正常细胞L-02也有明显的细胞毒性,在很低的烯二炔浓度下,就产生了强烈的抑制能力。为此在后续的工作中,可以围绕以下几个方面展开:第一,烯二炔3整个分子的疏水性比较强,加之整个细胞的微环境是亲水性的,使得烯二炔3进入细胞的难度加大。可以将烯二炔3的封端苄基直接替换成其他亲水性比较好的基团,增加整个分子的亲水性,亦或是将惰性的苄基替换成其他活性基团,可以进一步与亲水性的功能化基团反应。这样既可能增加烯二炔3的抗肿瘤毒性,也能使烯二炔3分子兼具其他功能。第二,肿瘤细胞和正常细胞微环境的差异为我们解决烯二炔3的细胞选择性提供了一条思路。比如根据肿瘤细胞和正常细胞微环境酸性的不同,我们可以改变烯二炔3分子的炔基取代基,使其成为pH响应的功能化药物小分子。另外也可以在烯二炔3分子的炔基末端修饰光响应型的基团,当药物分子释放完毕后,局部照射肿瘤部位引发烯二炔的活性。这样就可以靶向作用肿瘤细胞,在保持烯二炔3强烈的抗肿瘤细胞增殖能力的同时,大大减少对正常细胞的细胞毒性。第三,烯二炔3也可以负载在具有肿瘤靶向的载体上,传输到肿瘤特定区域脱附载体后,释放烯二炔的抗肿瘤活性。

图6 不同浓度烯二炔3对肿瘤和正常细胞的细胞毒性
3 结论与展望
本文从烯二炔抗生素的合成着手,设计并合成了基于马来酰亚胺为主体结构的烯二炔。马来酰亚胺部分羰基的吸电子效应大大降低Bergman环化反应的热引发温度。该烯二炔的Bergman环化反应得到多种谱学测试的证实。细胞毒性证明其可以对肿瘤细胞产生高效的抑制作用,其IC50可以媲美商用抗肿瘤药物。但是,该种烯二炔对正常细胞也显示出了强烈的抗增殖能力,说明它缺乏细胞选择性。这可以通过在烯二炔上引入肿瘤靶向基团,或者将该种烯二炔负载在具有肿瘤靶向的载体上解决。无论如何,该种烯二炔合成路线简单,细胞毒性作用高效,为以后设计合成高选择性、高效率开环烯二炔抗生素提供了一个新的思路。
[1]JEMAL A,BRAY F,CENTER M M,etal.Global cancer statistics[J].CA:A Cancer Journal for Clinicians,2011,61(2):69-90.
[2]ISHIDA N,MIYAZAKI K,KUMAGAI K,etal.Neocarzinostatin,antitumor antibiotic of high molecular weight;isolation,physicochemical properties,and biological activities[J].J Antibiot:Ser A,1965,18(2):68-76.
[3]EDO K,MIZUGAKI M,KOIDE Y,etal.The structure of neocarzinostatin chromophore possessing a novel bicyclo-[7,3,0] dodeca diyne system[J].Tetrahedron Letters,1985,26(3):331-334.
[4]GOLIK J,DUBAY G,GROENEWOLD G,etal.Esperamicins,a novel class of potent antitumor antibiotics:3.Structures of esperamicins A1,A2,and A1b[J].Journal of the American Chemical Society,1987,109(11):3462-3464.
[5]LEE M D,DUNNE T S,CHANG C C,etal.Calichemicins,a novel family of antitumor antibiotics:2.Chemistry and structure of calichemicin gamma.1I[J].Journal of the American Chemical Society,1987,109(11):3466-3468.
[6]KONISHI M,OHKUMA H,MATSUMOTO K,etal.Dynemicins,new antibiotics with the 1,5-diyn-3-ene and anthraquinone subunit:I.Productin,isolation and physico-chemical properties[J].The Journal of Antibiotics,1991,44(12):1300-1305.
[7]LEET J E,SCHROEDER D R,HOFSTEAD S J,etal.Kedarcidin,a new chromoprotein antitumor antibiotic:Structure elucidation of kedarcidin chromophore[J].Journal of the American Chemical Society,1992,114(20):7946-7948.
[8]KEN-ICHIRO Y,MINAMI Y,AZUMA R,etal.Structure and cycloaromatization of a novel enediyne,C-1027 chromophore[J].Tetrahedron Letters,1993,34(16):2637-2640.
[9]GALM U,HAGER M H,VAN LANEN S G,etal.Antitumor antibiotics:Bleomycin,enediynes,and mitomycin[J].Chemical Reviews,2005,105(2):739-758.
[10]SUGIMOTO Y,OTANI T,OIE S,etal.Mechanism of action of a new macromolecular antitumor antibiotic,C-1027[J].The Journal of Antibiotics,1990,43(4):417-421.
[11]ZHEN Y S,MING X Y,YU B,etal.A new macromolecular antitumor antibiotic,C-1027:Ⅲ.Antitumor activity[J].The Journal of Antibiotics,1989,42(8):1294-1298.
[12]SHEN B,LIU W,NONAKA K.Enediyne natural products:Biosynthesis and prospect towards engineering novel antitumor agents[J].Current Medicinal Chemistry,2003,10(21):2317-2325.
[14]ROWINSKY E K,EISENHAUER E A,CHAUDHRY V,etal.Clinical toxicities encountered with paclitaxel(Taxol)[J].Seminars in Oncology,1993,20(4,Suppl 3):1-15.
[15]SWAIN S M,WHALEY F S,EWER M S.Congestive heart failure in patients treated with doxorubicin[J].Cancer,2003,97(11):2869-2879.
[16]KAR M,BASAK A.Design,synthesis,and biological activity of unnatural enediynes and related analogues equipped with ph-dependent or phototriggering devices[J].Chemical Reviews,2007,107(7):2861-2890.
[17]BASAK A,KAR M.Benzofused N-substituted cyclic enediynes:Activation and DNA-cleavage potential[J].Bioorganic & Medicinal Chemistry,2008,16(8):4532-4537.
[18]JONES G B,LIN Y,MA D,etal.Congeners of the enediyne neocarzinostatin chromophore:Designed agents for bulged nucleic acid targets[J].Current Topics in Medicinal Chemistry,2008,8(6):436-447.
[19]SHARMA M,JOSHI M C,KUMAR V,etal.Synthesis and anticancer activity of 13-membered cyclic enediynes[J].Archiv Der Pharmazie,2011,344(9):564-571.
[20]POLUKHTINE A,KARPOV G,POPIK V V.Towards photoswitchable enediyne antibiotics:Single and two-photon triggering of Bergman cyclization[J].Current Topics in Medicinal Chemistry,2008,8(6):460-469.
[21]SONG D,SUN S,TIAN Y,etal.Maleimide-based acyclic enediyne for efficient DNA-cleavage and tumor cell suppression[J].Journal of Materials Chemistry B,2015,3(16):3195-3200.
[22]SONG D,TIAN Y,HUANG S,etal.An acyclic enediyne with a furyl tethering group for efficient inhibition of tumor cell viability[J].Journal of Materials Chemistry B,2015,3(43):8584-8588.
[23]SUN S,ZHU C,SONG D,etal.Preparation of conjugated polyphenylenes from maleimide-based enediynes through thermal-triggered Bergman cyclization polymerization[J].Polymer Chemistry,2014,5(4):1241-1247.
[24]DUBERNET M,CAUBERT V,GUILLARD J,etal.Synthesis of substituted bis(heteroaryl)maleimides[J].Tetrahedron,2005,61(19):4585-4593.
[25]SOUFFRIN A,CROIX C,VIAUD-MASSUARD M C.Efficient and general protocol for sonogashira cross-coupling reactions of maleimides[J].European Journal of Organic Chemistry,2012,2012(13):2499-2502.
[26]USUKI T,KAWAI M,NAKANISHI K,etal.Calicheamicin gamma(I)(1) and phenyl tert-butyl nitrone(PBN):Observation of a kinetic isotope effect by an ESR study[J].Chemical Communications,2010,46(5):737-739.
[27]NICOLAOU K C,OGAWA Y,ZUCCARELLO G,etal.Cyclic conjugated enediynes related to calicheamicins and esperamicins:Calculations,synthesis,and properties[J].Journal of the American Chemical Society,1988,110(14):4866-4868.
[28]SCHREINER P R.Monocyclic enediynes:Relationships between ring sizes,alkyne carbon distances,cyclization barriers,and hydrogen abstraction reactions:Singlet-triplet separations of methyl-substitutedp-benzynes[J].Journal of the American Chemical Society,1998,120(17):4184-4190.
[29]SANTOS J C,ANDRES J,AIZMAN A,etal.A theoretical study on the reaction mechanism for the bergman cyclization from the perspective of the electron localization function and catastrophe theory[J].Journal of Physical Chemistry A,2005,109(16):3687-3693.
[30]HABERHAUER G,GLEITER R,FABIG S.Enediyne dimerization vs bergman cyclization[J].Organic Letters,2015,17(6):1425-1428.
[31]MIFSUD N,MELLON V,PERERA K P U,etal.In situ EPR spectroscopy of aromatic diyne cyclopolymerization[J].Journal of Organic Chemistry,2004,69(18):6124-6127.
[32]USUKI T,NAKANISHI K,ELLESTAD G A.Spin-trapping of thep-benzyne intermediates from ten-membered enediyne calicheamicin gamma1I[J].Organic Letters,2006,8(24):5461-5463.
[33]ABE M.Diradicals[J].Chemical Reviews,2013,113(9):7011-7088.
[34]FALLE H R,LUCKHURST G R,LEMAIRE H,etal.The electron resonance of ground state triplets in liquid crystal solutions[J].Molecular Physics,1966,11:49-56.
Design,Synthesis and Cytotoxicity Study of a Novel Acyclic Enediyne
HUANG Shuai, LI Jing, LI Bao-jun, HU Ai-guo
(Shanghai Key Laboratory of Advanced Polymeric Materials,School of Materials Science and Engineering,East China University of Science and Technology,Shanghai 200237,China)
An acyclic enediyne with maleimide as the main structure and two symmetrical alkynyl chains was synthesized through Sonogashira coupling reaction.The enediyne was subjected to thermal Bergman cyclization to yield diradical intermediates.FT-IR results showed the complete conversion of the alkynyl moiety.The diradical intermediates could be further trapped by a spin trap (PBN) to generate mono- and di-PBN adducts corroborating the occurrence of the Bergman cyclization,which was confirmed by EPR analysis.MTT cell activity experiments confirmed that the half inhibitory concentration of this small molecular enediyne on tumor cell was around 18 μmol/L which were stable enough for long-time storage and active enough to suppress the cell multiplication for potential clinical application.
enediyne; Bergman cyclization; antibiotics; cytotoxicity
1006-3080(2017)01-0001-07
10.14135/j.cnki.1006-3080.2017.01.001
2016-04-19
国家自然科学基金项目(21474027)
黄 帅(1990-),男,硕士生,主要从事有机小分子合成研究。E-mail:hsecust@163.com
胡爱国,E-mail:hagmhsn@ecust.edu.cn
O621.3
A
猜你喜欢
中老年保健(2022年5期)2022-08-24
中老年保健(2021年6期)2021-08-24
当代水产(2021年6期)2021-08-13
学生天地(2020年14期)2020-08-25
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
小哥白尼(野生动物)(2019年5期)2019-08-27
中成药(2018年11期)2018-11-24
科学中国人(2018年8期)2018-07-23
中国生化药物杂志(2015年4期)2015-07-07
