
MELAS1例病例报告☆
2017-03-06 01:37:16刘会佳宋秀娟王北平邓晓红崔萌张海宁于萍檀国军郭力
中国神经精神疾病杂志 2017年11期
刘会佳宋秀娟○☆王北平邓晓红崔萌张海宁于萍檀国军郭力
线粒体脑肌病伴高乳酸血症和卒中样发作(mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes,MELAS)是因线粒体DNA(mitochondrial DNA,mtDNA)或核DNA(nuclear DNA,nDNA)突变致线粒体结构和(或)酶功能障碍,ATP生成不足,临床表现为以中枢神经系统及肌肉组织功能受损为主的多系统功能障碍的母系遗传病。因临床表现多种多样,难以早期诊断。本文报告1例MELAS,对其临床资料进行探讨,总结早期诊断要点,以期增强临床医生对该病早期诊断的意识。
1 临床资料
1.1 发病情况
患者,男,47岁,主因头痛、头晕伴左上肢无力3 d入院。患者入院3 d前无明显诱因出现头痛、头晕,伴左上肢无力、言语不利、右侧口角流涎,不能自行行走,需家人搀扶。于当地医院就诊,头颅CT示右侧颞顶叶大面积脑梗死,诊断为脑梗死并予以治疗,症状未见明显好转遂就诊于我院。既往14年前无明显诱因出现双耳听力下降,9年前大量饮酒后双耳失聪;间断头痛病史6年;行走不稳病史2年;左上肢抽搐病史2年;否认其他疾病。患者父亲(Ⅰ2)曾患高血压,其母亲(Ⅰ1)、大姐(Ⅱ1)及哥哥(Ⅱ3)患有双耳失聪及脑梗死;二姐(Ⅱ2)患有脑出血;爱人(Ⅱ5)及孩子(Ⅲ1、Ⅲ2)体键(图1)。
1.2 体格检查患者生命体征平稳,身材矮小,双耳失聪,心肺腹查体未见明显异常。神经系统查体:神清,不语,反应迟顿,定向力查体欠合作。左侧肢体肌力Ⅳ+级,双上肢肌张力增高,感觉、共济查体欠合作,右侧Babinski’s征(+)性,左侧Babinski’s征(±)性,余神经系统查体未见明显异常。
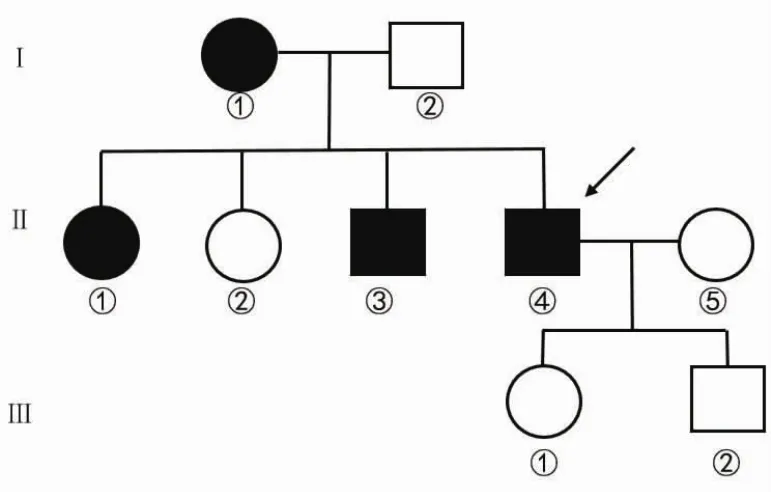
图1 家系图
1.3 辅助检查生化全项示:肌红蛋白204(0.00~85.00)ng/mL,葡萄糖6.58(3.90~6.10)mmol/L;尿常规示:酮体(+);血气分析示:PH 7.419(7.35~7.45),乳酸2.27 mmol/L;血常规、便常规、凝血常规均未见异常;心电图示:窦性心律,Ⅱ、Ⅲ、aVF、V4~V6 T波倒置、双向;视频脑电图示:中度异常。2017年2月10日行头颅MRI及磁共振波谱分析(MR spectroscopy,MRS)(图2 B)示:右侧大脑半球异常信号,考虑MELAS可能性大,颅内动脉MRA未见明显异常。2017年2月18日复查头颅MRI示:右侧大脑半球异常信号较前范围减少(图2 C);双侧额叶、半卵圆中心、放射冠散在缺血灶未见明显变化。因患者入院时不能自行行走,不语,未行乳酸运动试验。患者拒绝行肌肉活检,患者子女及兄姐拒绝行基因检测。MELAS综合征基因检测分析报告(2017年2月26日北京海思特临床检验所图3)示mtDNA A3243G突变。
1.4 诊断、治疗及随访入院后据相关辅助检查结果考虑MELAS可能性大,给予改善循环、营养脑神经、提供能量、改善脑代谢等综合治疗,后患者病情好转,可发单字音,可短距离自行行走。
2 讨论
MELAS于1984年由PAVLAKIS首次报告[1],是最常见的多系统功能障碍性线粒体病。多有以下临床表现:①母系遗传,本例患者母亲及大姐、哥哥均患有双耳失聪及脑梗死;②身材矮小,智能发育迟滞或痴呆,本例患者身高158 cm,体重55 kg,是一名语文老师,并无智能发育迟滞表现;③四肢近端极度不能耐受疲劳、神经性耳聋、头痛、癫痫、失语、视力受损及认知功能下降等,在本例患者表现为6年来间断头痛、双耳失聪等;④可伴随糖尿病、心肌病、肾病等[2]。辅助检查:头颅CT典型表现为双侧基底节区钙化(图1A);MRI可见与血管分布区不一致的累及皮层的病灶(图1B);MRS于感兴趣区内可见乳酸(lactate,Lac)峰(图1D);肌电图多呈肌源性损害;肌肉活检及改良Gomori染色见破碎红纤维(RRF),本例患者家属拒绝行肌电图及肌肉活检;运动试验后血乳酸升高;线粒体基因检测到突变基因等。由于本病表现多样,早期诊断困难,且常误诊为病毒性脑炎、多发性硬化、脑卒中等疾病,致患者反复发病,生存质量下降。因此,早期诊断MELAS至关重要,应注意以下几方面。
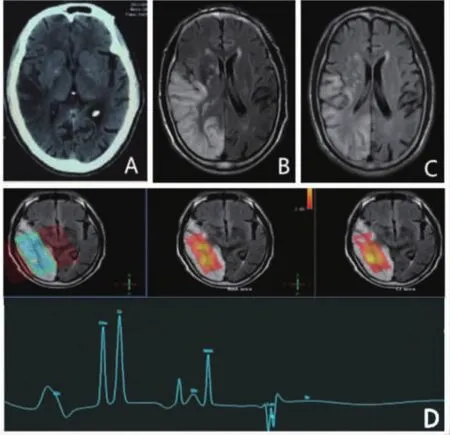
图2 头颅MRI及磁共振波谱分析A:双侧基底节区钙化;B:住院第5天行头颅MRI示右侧颞枕叶异常信号,与血管分布区域不一致;C:住院第14天头颅MRI示右侧大脑半球异常信号较前范围减少;D:箭头所示住院第5天MRS示感兴趣区内可检测到乳酸峰
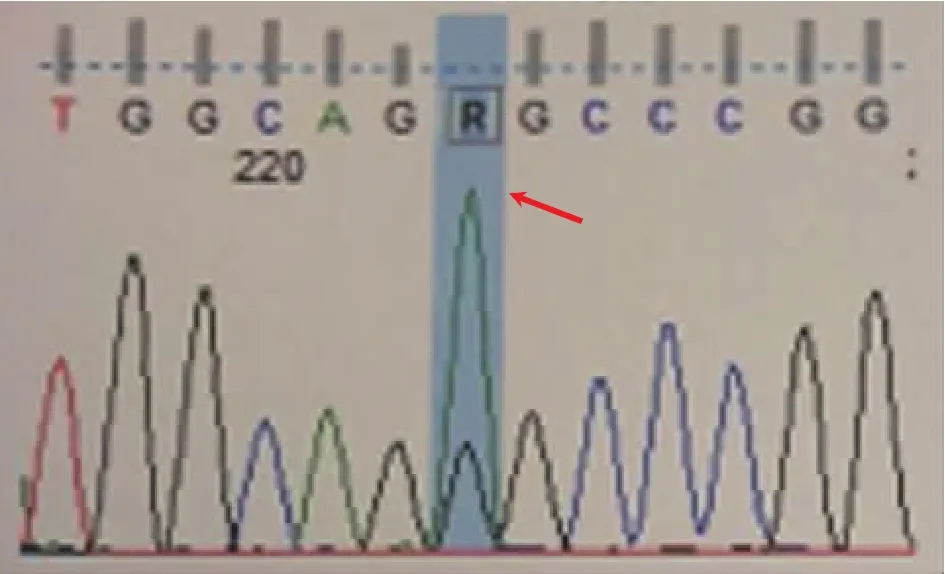
图3 基因检测分析箭头所示MELAS综合征基因检测分析示mtDNA A3243G突变。
首先,重视青年原因不明的双耳听力障碍。约30%~75%的MELAS患者早期出现严重的听力损伤[3],多为渐进性加重,也可伴随脑卒中样发作而突发或波动[4-5]。研究表明线粒体基因突变(如OPA1)可能与毛细胞选择性丧失有关,导致耳蜗功能障碍,听力受损[6]。
其次,评估脑组织代谢产物。MRS是一种无创的检测活体组织器官内代谢产物的技术。MELAS患者行MRS可见乳酸峰,N乙酰天门冬氨酸(N-acetyl-L-aspartate,NAA)/肌酸(Creatine,Cr)降低,Lac/Cr增高。当机体内细胞缺血、缺氧及线粒体功能障碍时氧化磷酸化受抑制,糖酵解水平升高[7-8],代谢产物乳酸生成增多,因此MELAS患者脑内病变区域可检测到乳酸峰,这与疾病严重程度相关[9]。NAA主要存在于神经元中,其合成减少间接反应神经元损伤、功能紊乱、缺失,或线粒体功能受损[10]。临床上通常检测感兴趣区内的脑代谢水平,但在看似正常的灰质中也可检测出乳酸峰,或许这更具有临床意义[8,11],本例患者尚未检测,应在今后的临床工作中加以应用。约80%的MELAS患者为编码亮氨酸的tRNA基因A3243G点突变致病(如:图3)[7],然而临床上对于A3243G基因携带者的早期预防关注不足。一项前瞻性研究表明A3243G基因携带者较非基因携带者有更高的灰质总胆碱(total choline,tcho)水平,此生物学标记物有望用于评估A3243G基因携带者疾病进展及MELAS患者疾病缓解程度[12]。
再次,检测脑组织血流灌注情况。传统观点认为MELAS患者病变区域为血流低灌注的,然而有研究表明,在卒中样发作发生之前,区域血流高灌注可持续3个月[13]。YeH等应用单光子发射计算机断层扫描(SPECT)研究表明:MELAS患者卒中样发作受损(stroke-like episodes lesion,SELs)区域的脑组织为高灌注区,但对示踪剂的摄取不足;与正常脑组织相比SELs周围区域摄取示踪剂有所增加[14]。其机制为急性损伤时发生氧化应激反应,超急性期脑血流量及糖代谢增加,亚急性期脑血流量减少及葡萄糖代谢降低,随后神经元损伤致脑血流量及糖利用率进一步下降[15]。此外,IKAWA等[13]应用动脉自旋标记(Arterial spin labeling,ASL)灌注核磁共振成像在静止期SELs区域检测到血流高灌注,与临床前潜在病变相对应,可出现在卒中样发作前数月。
综上所述,在临床工作中当患者具有听力受损的病史、不耐疲劳、神经系统功能受损等表现时,应进行有助于早期诊断MELAS的辅助检查:运动试验检测乳酸水平;MRS检测病灶区和(或)非病灶区代谢指标;SPECT等检测脑血流灌注等。确诊则需要行肌肉活检或基因型检测。未来随着我们对MELAS的认识不断深入,有望做到早期诊断,改善患者的生存质量。
[1]PAVLAKIS S,PHILLIPS PC,DIMAURO S,et al.Mitochondrial myopathy,encephalopathy,lacticacidosis,andstrokelike episodes:a distinctive clinical syndrome[J].Ann Neurol,1984,16(4):481-488.
[2]FANG W,HUANG CC,LEE CC,et al.Ophthalmologic manifestations in MELAS syndrome[J].Arch Neurol,1993,50(9):977-980.
[3]WARRICK P.D,WARDROP P,SIMA D.W.Sensorineural hearing loss in MELAS syndrome[J].Laryngol.Otol,1997,111(3):279-281.
[4]SUE CM,LIPSET LJ,CRIMMINS DS,et al.Cochlear origin of hearing loss in ELAS syndrome[J].Ann.Neurol,1998,43(3):350-359.
[5]KARKOS PD,WALDRON M,JOHNSON IJ,et al.The MELAS syndrome.Review of the literature:the role of the otologist[J].Clin Otolaryngol,2004,29(1):1-4.
[6]ROSAMARIA S,ELONA C,PIETRO S,et al.Reply:Both mitochondrial DNA and mitonuclear gene mutations cause hearing loss through cochlear dysfunction[J].Brain,2016,139(6):e34.
[7]BUCK LT,PAMENTER ME.Adaptive responses of vertebrate neurons to anoxia:matching supply to demand[J].Respir Physiol Neurobiol,2006,154(1-2):226-240.
[8]MOLLER HE,KURLEMANN G,PUTZLER M,et al.Magnetic resonance spectroscopy in patients with MELAS[J].Neurol Sci,2005,229–230:131-139
[9]KAUFMANN P,ENGELSTAD K,WEI Y,et al.Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype[J].Neurology,2011,77(22):1965-1971.
[10]BIRKEN DL,OLDENDORF WH.N-acetyl-L-aspartic acid:a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain[J].Neurosci Biobehav Rev,1989,13(1):23-31.
[11]BIANCHI MC, TOSETTI M, BATTINI R, et al. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance [J]. AJNR Am J Neuroradiol, 2003, 24(10): 1958-1966.
[12]WEIDUSCHAT N,KAUFMANN P,Mao X,et al.Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers[J].Neurology,2014,82(9):798-805.
[13]IKAWA M,YONEDA M,MURAMATSU T,et al.Detection of preclinically latent hyperperfusion due to stroke-like episodes by arterial spin-labeling perfusion MRI in MELAS patients[J].Mitochondrion,2013,13(6):676-680.
[14]YEH HL,CHEN YK,CHEN WH,et al.Perfusion status of the stroke-like lesion at the hyperacute stage in MELAS[J].Brain and Development,2013,35(2):158-164.
[15]GOODFELLOW JA,DANI K,STEWART W,et al.Mitochondrial myopathy,encephalopathy,lactic acidosis and stroke-like episodes:an important cause of stroke in young people[J].Postgrad Med,2012,88(1040):326-334.
猜你喜欢
家庭科学·新健康(2023年9期)2023-10-01 09:20:06
湘潮(上半月)(2022年8期)2022-12-12 03:45:28
家庭科学·新健康(2022年1期)2022-02-02 02:03:18
心电与循环(2021年4期)2021-11-29 02:41:56
紫禁城(2020年5期)2021-01-07 02:13:34
心电与循环(2020年3期)2020-06-18 13:43:12
大观(2018年8期)2018-01-23 18:02:37
家庭科学·新健康(2016年11期)2016-11-23 11:57:37
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
扬子江(2016年1期)2016-05-19 23:29:21
