
精氨酸缺陷型菌株发酵生产反式-4-羟脯氨酸
2017-02-15 05:37王晓姣张震宇孙付保于林
食品与发酵工业 2017年1期
王晓姣,张震宇,孙付保,于林
(江南大学 生物工程学院,江苏 无锡,214122)
精氨酸缺陷型菌株发酵生产反式-4-羟脯氨酸
王晓姣,张震宇*,孙付保*,于林
(江南大学 生物工程学院,江苏 无锡,214122)
为了获得高产反式-4-羟脯氨酸的菌株,基于大肠杆菌的代谢网络模型的指导,以大肠杆菌E.coliBL21(DE3)ΔputA为出发菌株,通过基因敲除技术成功敲除argB基因,阻断L-脯氨酸合成的前体物L-谷氨酸的分支代谢途径,增加L-脯氨酸合成的代谢流,构建了精氨酸缺陷型菌株E.coliBL21(DE3)ΔputAΔargB。同时转入表达质粒pUC19-proB2A-Ptrp2-hyp,该质粒含有突变基因proB2,该突变基因所编码的谷氨酸激酶受L-脯氨酸的反馈抑制作用显著降低。摇瓶发酵结果表明,在外源添加600 mg/LL-精氨酸时,该重组菌株产反式-4-羟脯氨酸的量达到312.67 mg/L,较菌株E.coliBL21(DE3)ΔputA/pUC19-proB2A-Ptrp2-hyp提高了25.29%。
大肠杆菌1;反式-4-羟脯氨酸2;基因敲除3;脯氨酸4
反式-4-羟脯氨酸(trans-4-hydroxyproline,Hyp)为亚氨基酸,是L-脯氨酸经反式-4-羟化酶(proline-4-hydroxylase,hyp) 羟基化的产物,归属高附加值小品种氨基酸类[1]。Hyp在食品营养、化工生产、护肤美容业以及医药保健具有广泛的应用[2-3]。由于其潜在的医药、食品价值以及市场需求,促进了反式-4-羟脯氨酸的研究步伐。
脯氨酸作为羟脯氨酸合成的底物,前期实验室生产合成羟脯氨酸都需要外源添加脯氨酸[4-5]。脯氨酸的添加不仅导致生物法合成羟脯氨酸的成本大大提高,而且外源添加的脯氨酸大部分并不能被微生物所利用,造成了资源的浪费。
基于对代谢调节的认识,可以在基因水平和酶学水平上对Hyp的合成途径进行理性操作。目的产物的积累一般有2种方式,积累关键前体和降低副产物生成:L-谷氨酸是L-脯氨酸生物合成的前体物质,同时也是L-精氨酸、L-鸟氨酸和L-瓜氨酸生物合成的前体物质,其胞内浓度决定了L-脯氨酸的合成速率[6]。因此,削弱竞争代谢途径,阻断L-谷氨酸到L-精氨酸的合成代谢流,使得L-谷氨酸到L-脯氨酸合成分支代谢流量增强,使得碳源、氮源等更多的用于脯氨酸的合成,可以提高L-脯氨酸的产量[7]。谷氨酸是脯氨酸和精氨酸的共同前体物(图1)。为提高脯氨酸的产量可以采用基因敲除等方法切断谷氨酸合成精氨酸的途径,从而为脯氨酸的生产提供更多的原料。N-乙酰谷氨酸激酶(N-Acetylglutamate kinase EC 7 2.8,NAGK),是精氨酸合成途径的第2个酶。该酶由argB基因编码,是精氨酸途径中的关键酶。该酶不仅被精氨酸反馈抑制而且也被其反馈阻遏[8-9],而且是精氨酸生产过程中被精氨酸抑制的唯一限速酶。本文选择敲除精氨酸代谢途径中的argB基因,切断精氨酸代谢途径,使得脯氨酸大量积累,从而获得羟脯氨酸高产菌株。

图1 大肠杆菌中反式-4-羟脯氨酸生物合成途径Fig.1 The biosynthetic pathways of Hyp in E. coli
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
本实验用到的菌株[5]及质粒[10]见表1,其中E.coliBL21(DE3)作为宿主,质粒pUC19作为基因表达载体。

表1 实验所用菌株和质粒
1.1.2 试剂及仪器
葡萄糖、NaCl、MgSO4、K2HPO4、(NH4)2SO4、琼脂粉、CaCl2、FeSO4、正丙醇、氯胺T、高氯酸、对二甲氨基苯甲醛、异丙醇、NH4Cl等购自上海国药。胰蛋白胨、酵母抽提物、琼脂糖、氨苄青霉素钠、硫酸卡那霉素、氯霉素、TaqDNA聚合酶、PfuDNA 聚合酶、dNTP Mixture、SanPrep柱式质粒DNA小量抽提试剂盒等购自生工生物工程(上海)股份有限公司;DNA限制性内切酶,DNA Marker,购自宝生物公司(中国,大连)。
主要仪器包括培养箱、恒温摇床、分光光度计、电转仪、PCR仪、电泳仪、冷冻离心机、氨基酸分析仪等。
1.1.3 培养基及培养条件
LB培养基(胰蛋白胨10 g/L,酵母提取物5 g/L,NaCl 10 g/L,调pH至 7.0~7.2。固体培养基另加1.5%的琼脂粉。)用于培养E.coliBL21(DE3)。培养温度37 ℃。发酵培养基(葡萄糖10 g/L,甘油5 g/L,胰蛋白胨15 g/L,(NH4)2SO45 g/L,K2HPO43 g/L,FeSO43 mmol/L,MgSO40.2 g/L,CaCl20.015 g/L)用于反式-4-羟脯氨酸发酵,用NaOH调节 pH 至8.0,250 mL三角锥形瓶分装30 mL。菌株E.coliBL21(DE3)ΔputA/pUC19-proB2A-Ptrp2-hyp发酵生产时,需要添加6~10 g/LL-脯氨酸;E.coliBL21(DE3)ΔputAΔargB/pUC19-proB2A-Ptrp2-hyp初步发酵添加1 g/LL-精氨酸。
抗生素的工作质量浓度:氨苄青霉素50 μg/mL,卡那霉素50 μg/mL,氯霉素50 μg/mL。
1.1.4 引物设计
本实验所用引物序列见表2,由上海生工合成。

表2 实验所用引物
注:下划线部分是质粒pKD4的抗性基因序列。
1.2 重组大肠杆菌的构建
1.2.1 敲除基因argB获得精氨酸缺陷型菌株
利用Red/ET同源重组系统[11-13]进行基因敲除(图2)。
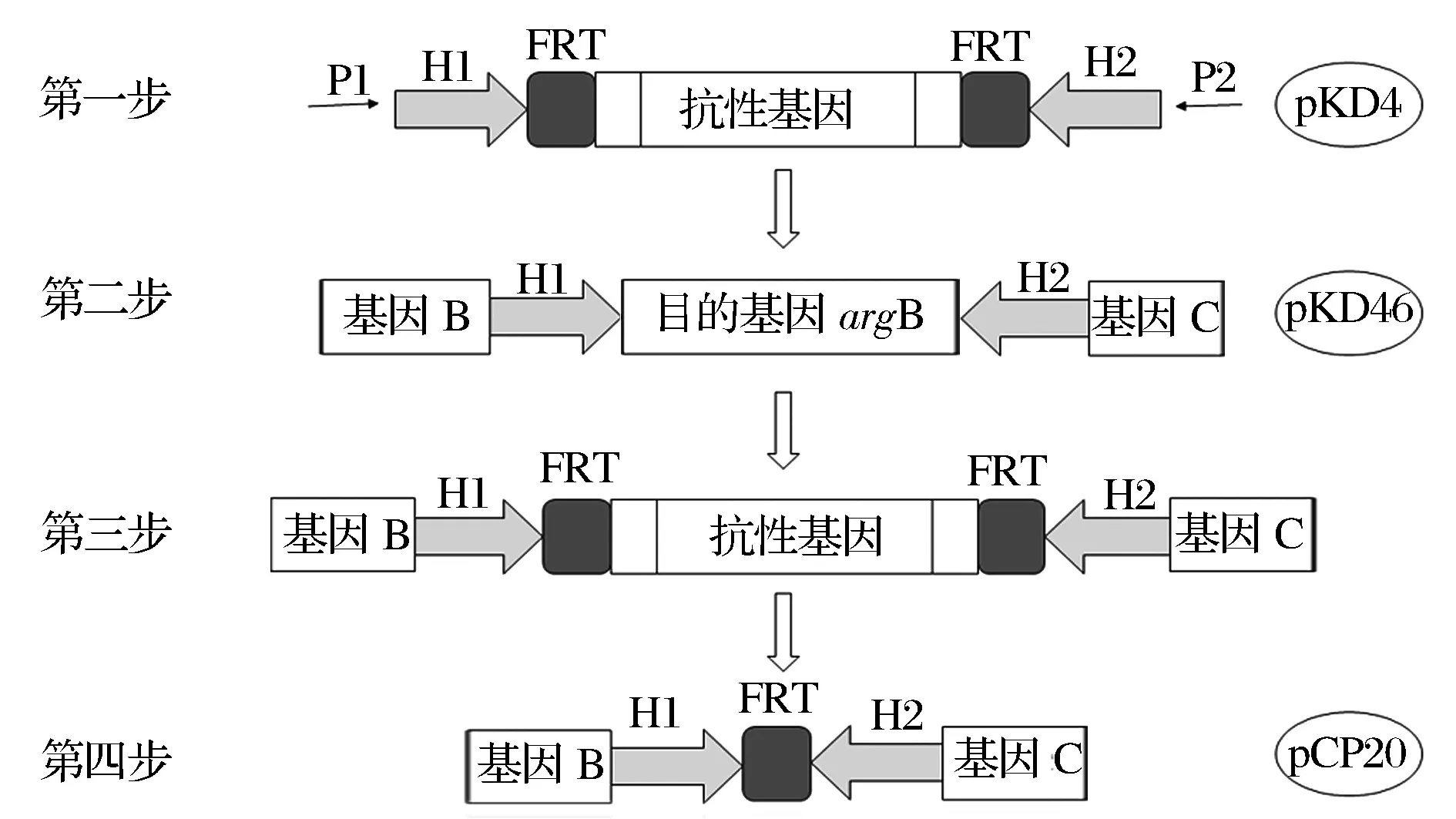
H2, H1是同源臂;P1, P2是抗性基因序列图2 利用Red/ET同源重组系统进行基因敲除的过程Fig.2 Processes of gene knockout using the plasmid-assistant Red homologous recombination
以辅助质粒pKD4为模板,利用引物P1、P2通过PCR扩增直接获得含有目的基因上下游同源臂和kan基因的线性打靶片段;将表达Red重组酶的辅助质粒pKD46导入E.coliBL21(DE3)ΔputA感受态细胞内,并制备为电转感受态细胞(制备过程中需要添加L-阿拉伯糖以诱导质粒pKD46的Exo蛋白、Beta蛋白和Gam蛋白的表达);将线性的打靶片段电转导入电转感受态细胞中以实现打靶片段与目的基因argB的配对重组(以kan基因片段替代目的基因argB),之后采用42 ℃热激耦合抗生素抗性负筛选的策略去除质粒pKD46;通过菌落PCR对在卡那抗性平板上筛到的单菌落进行验证;将表达FLP重组酶的辅助质粒pCP20导入菌落PCR验证正确的阳性转化子中,FLP重组酶可以直接作用于两个FRT位点并发生同源重组,去除2个FRT位点之间的kan基因片段以消除卡那抗性,并留下一个FRT“疤痕”[14]。
1.2.2 大肠杆菌电转感受态制备方法
挑取含有pkD46的E.coliBL21(DE3)ΔputA单菌落接种于氨苄抗性的新鲜LB液体培养基中,30 ℃,220 r/min过夜培养;取过夜种子液按1%接种量接种到50 mL 氨苄抗性的新LB培养基中,并加入4.8 g/L的L-阿拉伯糖,30 ℃,220 r/min振荡培养至OD600值达到0.5~0.6;将培养液分装于灭菌的50 mL离心管中,冰上预冷 10 min,4 ℃,4 000 r/min离心10 min,弃去上清液,回收细胞;加入等体积的冰预冷10%(v/v)甘油重悬,4 ℃,4 000 r/min离心10 min,重复甘油洗2次;小心弃去上清,利用残留的甘油重悬细胞,每管分装80 μL,-80 ℃保存。
1.2.3 大肠杆菌化转感受态制备方法
接种大肠杆菌单菌落于50 mL pH 7.0的新鲜LB液体培养基中,30 ℃,220 r/min过夜培养;取过夜种子液按1%接种量接种到pH 7.0的新鲜LB培养基中,30 ℃,220 r/min振荡培养(约2 h)至 OD600值达到0.5~0.6;将培养液分装于灭菌的50 mL离心管中,冰上预冷 10 min;4 ℃,1 000×g 离心5 min,弃去上清,回收细胞;加入原培养液5%体积的TSB培养基充分悬浮菌体,缓慢吹打均匀;冰上放置10 min后分装至已灭菌的1.5 mL离心管中,每管分装50 μL,-80 ℃保存。
1.2.4 大肠杆菌感受态细胞的转化方法(热击法)[4]
转化体系为:30 μL ddH2O,10 μL 5×KCM(5 mol/L KCl,0.15 mol/L CaCl2,0.25 mol/L MgCl2),10 μL重组质粒,10 μL感受态细胞。迅速混匀,先冰浴30 min;然后42 ℃热击90 s;接着冰浴5 min;最后加入600 μL新鲜的LB液体培养基,37 ℃ 220 r/min振荡培养1 h;取100 μL复苏培养后的菌液涂布于含抗生素的LB平板,于37 ℃恒温培养箱中培养8~16 h。
1.3 发酵试验
1.3.1 摇瓶发酵
将重组大肠杆菌E.coliBL21(DE3)ΔputAΔargB/ pUC19-BH在含有氨苄霉素的LB平板上划线培养;挑取单菌落接种于含有50 μg/mL 氨苄的LB液体培养基中(30 mL/250 mL三角瓶),37 ℃ 220 r/min振荡培养8 h,作为种子液;按6%接种量取上述种子培养液1.8 mL接入发酵培养基(30 mL/250 mL三角瓶),30 ℃ 220 r/min振荡培养24 h。
1.3.2 生物量的测定
取发酵液,用去离子水稀释相应倍数,并以去离子水为空白对照,测定600 nm波长处的吸光度值。
1.3.3 葡萄糖的测定
取1 mL发酵液,8 000 r/min 离心2 min,取200 μL上清液,加入200 μL DNS试剂,沸水浴5 min;用冷水冷却样品,然后加2.6 mL去离子水稀释到3 mL,用分光光度计在540 nm波长处测定吸光度值。
1.3.4L-脯氨酸的含量测定
L-脯氨酸的检测采用酸性茚三酮显色法:将发酵液离心后,取上清,适当稀释后取1 mL于10 mL试管中,加入1 mL冰乙酸;摇匀后加入1 mL酸性茚三酮;摇匀后沸水浴1 h;接着加入冰乙酸2 mL,摇匀后冷水冷却,测定515 nm波长处的吸光度值。
1.3.5 反式-4-羟脯氨酸积累量的测定
反式-4-羟脯氨酸的检测采用氯胺T法:将发酵液离心后,取上清,稀释后取2.5 mL于10 mL试管中,加入1 mL氯胺T,摇匀后室温放置20 min;然后加入1 mL显色剂,摇匀后迅速将试管置于60 ℃水浴锅中,20 min后取出冷水冷却,用分光光度计在560 nm波长处测定吸光度值。
1.3.6 反式-4-羟化酶酶活的测定[15]
全细胞酶活测定条件:测定发酵液的OD600,根据大肠杆菌OD600和细胞干重的常用转换计算公式,根据DCW(g/L) =0.54×OD600测得细胞干重DCW。发酵液12 000 r/min离心2 min后,弃掉上清液,回收菌体,将500 μL的酶反应缓冲液(MES 240 mmol/L(pH 6.5),脯氨酸20 mmol/L,2-酮戊二酸40 mmol/L,硫酸亚铁4 mmol/L,维生素C 8 mmol/L)加入到称重好的菌体中,用酶反应缓冲液将细胞重新悬浮后,在35 ℃摇床中,220 r/mim培养15 min后,转移到100 ℃水浴中加热2 min终止酶反应,并测定反式-4-羟脯氨酸浓度,反式-4-羟脯氨酸的测定方法同上1.3.5。1个单位酶活定义1 min将1 nmol脯氨酸完全转化为反式-4-羟脯氨酸的酶量,单位为U,全细胞酶活是每1 mg干菌体的酶活,单位为U/mg。
1.3.7 乙酰谷氨酸激酶活力测定[16]
2 mL反应总体积中,含0.1 mL 酶液,0.1 mL 0.2 mol/L盐酸羟胺,0.28 mL 0.14 mol N-乙酰谷氨酸,0.2 mL 0.02 mol MgCl2,0.14 mL 0.014 mol ATP,1.18 mL 0.1 mol Tris-HCl缓冲液(pH8.0),37 ℃水浴保温30 min,加2 mL FeCl3试剂(3.4%FeCl3溶于2 mol/L HCl,并与8%三氯乙酸等体积混匀)终止反应,测OD540值。1个酶活力单位(U)是指在上述条件下,反应体系中每分钟催化1 μmol N-乙酰谷氨酸氧化所需的酶量。
1.3.8 氨基酸的测定
发酵液经12 000 r/min 离心5 min 后,取上清500 μL,加入500 μL 10%磺基水杨酸沉淀4 h,用0.02 mol/L HCl稀释适当倍数,用0. 22 μm微孔膜过滤后,采用氨基酸分析仪 L-8900测定氨基酸。
2 结果与分析
2.1argB敲除菌株的验证
经过抗性平板筛选,挑出只在非抗性平板上菌落,用验证引物Y1、Y2进行菌落PCR,并进行核酸胶验证。筛选到argB成功敲出的菌株。从图3中可以看到出发菌株E.coliBL21(DE3)ΔputA菌落PCR得到片段大小为1 000 bp,与通过验证引物Y1、Y2进行菌落PCR获得的片段大小相当(片段大小为964 bp);卡那片段成功替换菌株菌落PCR得到片段大小为1 700 bp左右,与卡那基因片段大小相当;消抗后从图3可以看到除了菌株3-8,其余15株菌经过菌落PCR得到大小为300 bp左右的片段,说明argB基因被成功敲除。由于利用Red/ET同源重组系统进行基因敲除会留下一个FRT“疤痕”,可能会对菌株生长造成影响,因此,本实验对该15株菌进行了初步发酵,发现菌株3-4脯氨酸产量高于其他菌株,且高于菌株BL21(DE3)ΔputA(图4),得到菌株BL21(DE3)ΔputAΔargB。

M-5 000 bp DNA Ladder Marker;1-E. coliBL21(DE3) ΔputA的菌落PCR结果;2-E. coli BL21(DE3) ΔputA抗性替换成功菌株的菌落PCR结果;3-抗性消除菌株的菌落PCR结果图3 argB基因敲除菌株的PCR鉴定Fig.3 PCR identification of argB gene knockout strain
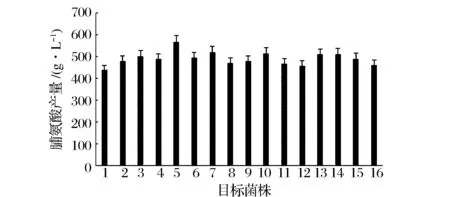
1代表菌株BL21(DE3)ΔputA的初步发酵结果;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16分别代表目标菌株 3-1、3-2、3-3、3-4、3-5、3-6、3-7、3-9、3-10、3-11、3-12、3-13、3-14、3-15、3-16的初步发酵结果图4 argB基因敲除菌株的初步发酵Fig.4 The preliminary fermentation of argB gene knockout strain
2.2 重组菌株的构建
通过材料与方法1.2.4中化学转化的方法,构建菌株BL21(DE3)ΔputAΔargB/pUC19-BH。通过使用SanPrep柱式质粒DNA小量抽提试剂盒提取重组菌株BL21(DE3)ΔputAΔargB/pUC19-BH。质粒进行单双酶切验证(图5)。质粒通过BamH I、KpnI进行双酶切,分别获得基因片段hyp-Ptrp2-proB2A(BH片段,3416 bp)、载体pUC19(2 686 bp),结果如泳道1;表达载体通过BamH I进行单酶切,重组质粒的大小约6 102 bp,结果如泳道2;从图5可以看出这些片段的大小均与预期目标相符,表明重组菌株构建成功。

M1-5000bp DNA Ladder Marker;M2-5000bp DNA Ladder Marker;1-双酶切结果;2-单酶切结果图5 表达质粒酶切电泳验证Fig.5 The electrophoresis test and verify of recombinant plasmid pUC19-BH by enzyme digestion
2.3 氨基酸分析仪检测发酵液中氨基酸成分
通过氨基酸分析仪进行初步定性分析结果(图6),可以发现菌株BL21(DE3)ΔputA/pUC19-BH与菌株BL21(DE3)ΔputAΔargB/pUC19-BH发酵结果相比,前者精氨酸的含量明显高于后者,精氨酸缺陷型菌株几乎没有精氨酸积累,这进一步证明后者精氨酸途径被成功切断。同时,我们看到发酵液中有反式-4-羟脯氨酸生成,说明羟化酶得到表达,菌株BL21(DE3)ΔputAΔargB/pUC19-BH构建成功。
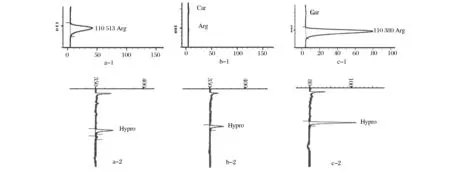
图6 发酵液中各氨基酸分析结果Fig.6 The analytical results of amino acids in fermentation broth图a-1,a-2:菌株BL21(DE3)ΔputA/pUC19-BH发酵液中氨基酸分析结果;图b-1,b-2:无外源添加L-精氨酸时,菌株BL21(DE3)ΔputAΔargB/pUC19-BH发酵液中氨基酸分析结果;图c-1,c-2:外源添加适量L-精氨酸时,菌株BL21(DE3)ΔputAΔargB/pUC19-BH与发酵液中氨基酸分析结果
2.4argB基因敲除菌株发酵参数
对敲除菌进行初步发酵,并测定菌株的发酵参数,从结果(图7)中,我们发现精氨酸缺陷型菌株无论是生长状况,葡萄糖消耗还是产反式-4-羟脯氨酸的能力均受到一定抑制。对于argB基因敲除菌株,该基因编码的N-乙酰谷氨酸激酶的酶活几乎为0 mU/mg,这更进一步说明argB基因被成功敲除。另外,研究发现,当培养基中外源添加1 g/L的L-精氨酸时,菌体的生长状况得到了恢复,且反式-4-羟脯氨酸的产量与出发菌株相比有所提高。因此,后文L-我们对精氨酸的添加量进行了研究。

a-菌株BL21(DE3)ΔputA/pUC19-BH的发酵参数;b-无外源添加L-精氨酸时,菌株 BL21(DE3)ΔputAΔargB/pUC19-BH的发酵参数;c-外源添加1 g/L L-精氨酸时,菌株BL21(DE3)ΔputAΔargB/pUC19-BH的发酵参数图7 argB基因敲除菌株发酵结果Fig.7 The fermentationresults of argB knockout strains
2.5L-精氨酸对BL21(DE3)ΔputAΔargB/pUC19-BH反式-4-羟脯氨酸产量的影响
由于菌株是L-精氨酸营养缺陷型,所以种子培养基中需要加入适量的精氨酸来满足菌体生长的需求。本实验研究了精氨酸不同浓度对菌体的生长。
argB基因的敲除,使得精氨酸合成受阻,菌株变为L-精氨酸缺陷型菌株,因此外源添加精氨酸,可以补充菌体生命活动所需的物质。BL21(DE3)ΔputAΔargB/pUC19-BH菌体生长能力的恢复,有利于反式-4-羟脯氨酸产量的进一步提高。发酵培养基中添加不同浓度的精氨酸,观察菌体的生长及反式-4-羟脯氨酸产量情况,结果如图8所示。随着精氨酸浓度的增加,菌株的生长及反式-4-羟脯氨酸合成能力均有所提高。当发酵培养基中添加的L-精氨酸浓度达到600 mg/L后,随着精氨酸添加量的增加,反式-4-羟脯氨酸产量增长趋势变小。从生产成本考虑,选择在培养基中添加600 mg/L的L-精氨酸,此时反式-4-羟脯氨酸产量达到 312.67 mg/L,较出发菌株BL21(DE3)ΔputA/pUC19-BH产量233.59 mg/L提高了25.29% ;细胞生长量也恢复到了亲本水平。由此可见,精氨酸的添加有利于菌株的生长及反式-4-羟脯氨酸的积累。
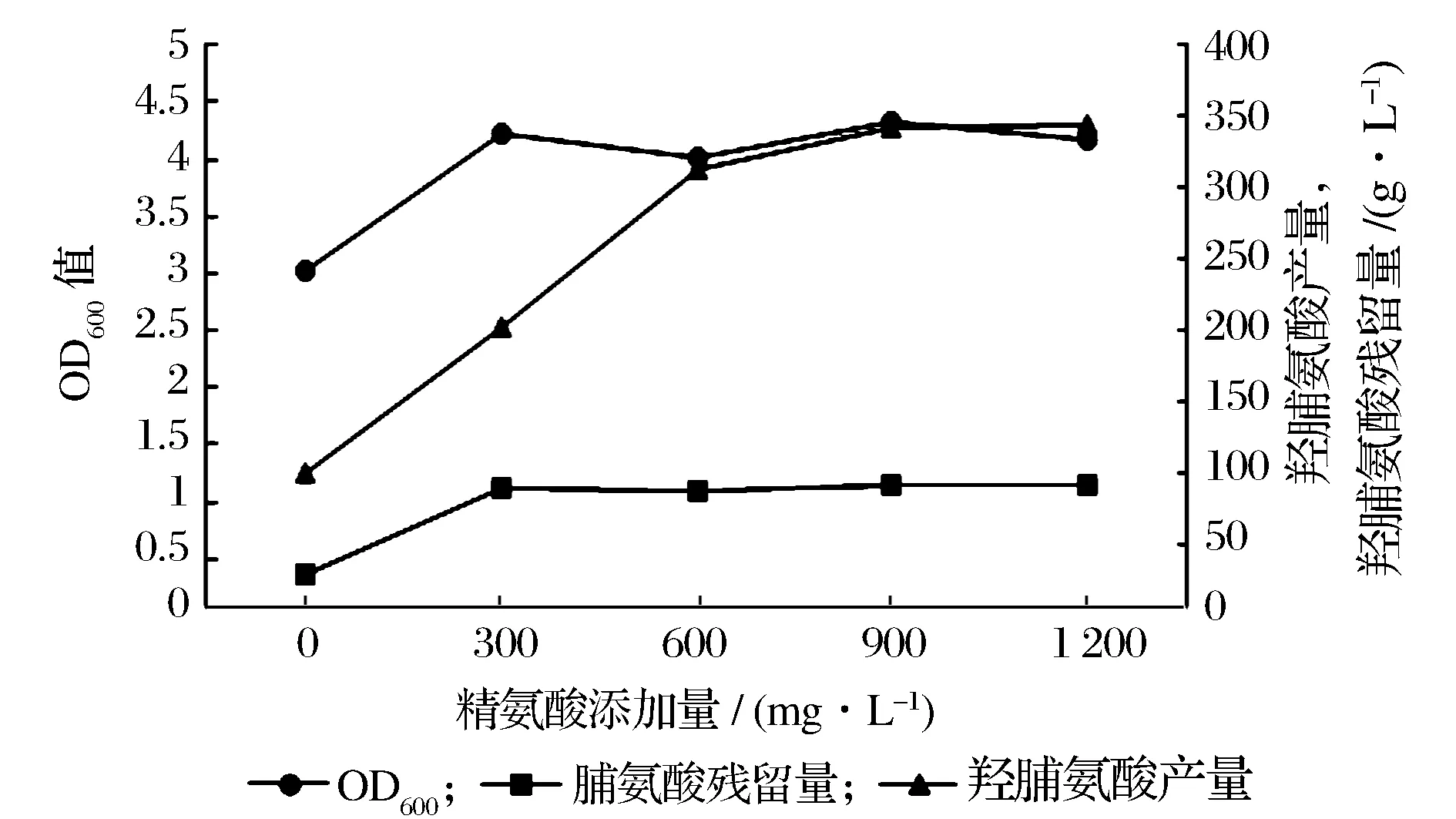
图8 L-精氨酸添加量的优化Fig.8 The optimization of addition amount of L-arginine
3 讨论
本研究通过Red/ET同源重组系统进行基因敲除,成功构建了argB基因敲除菌株 BL21(DE3)ΔputAΔargB,结果发现该重组菌株的反式-4-羟脯氨酸产量有所提高。说明argB基因的敲除有利于反式-4-羟脯氨酸的生产。
argB基因编码精氨酸合成途径的乙酰谷氨酸激酶。敲除argB基因后,胞内精氨酸合成受阻,可能使更多前体谷氨酸流向L-脯氨酸合成途径,从而提高作为反式-4-羟脯氨酸底物的L-脯氨酸产量。但L-精氨酸是必须氨基酸,对维持菌体正常生命活动有重要意义,所以其敲除影响了菌体的生长。我们对L-精氨酸的添加量进行了研究,结果发现适量的精氨酸添加可以使得敲除菌株恢复生长性状,同时反式-4-羟脯氨酸的产量达到312.67 mg/L与实验室原有水平相比有了显著的提高。
该生产反式-4-羟脯氨酸的方法与原生产方法相比,原生产法方在摇瓶发酵时需要添加6~10 g/L的L-脯氨酸作为底物,而该生产方法只需添加0.6~1 g/L的L-精氨酸来满足菌体生长;且L-脯氨酸的价格高于L-精氨酸。因此本文所构建的菌株在生产过程中展现出成本低,发酵过程易控制,发酵水平稳定等优势,这为反式-4-羟脯氨酸的工业化生产奠定了良好基础。
[1] VICKERY H B, SCHMIDT C L A. The history of the discovery of the amino acids[J]. Chemical Reviews,1931,9(2): 169-318.
[2] BRANDS K M J, JOBSON R B, CONRAD K M, et al. Efficient one-pot synthesis of the 2-aminocarbonylpyrrolidin-4-ylthio-containing side chain of the new broad-spectrum carbapenem antibioticertapenem[J].The Journal of Organic Chemistry,2002,67(14): 47 71-4 776.
[3] PHANG J M, DONALD S P, PANDHARE J, et al. The metabolism of proline, a stress substrate, modulates carcinogenic pathways[J]. Amino Acids, 2008, 35(4): 681-690.
[4] 刘合栋. 高产反式-4-羟脯氨酸重组大肠杆菌的构建和发酵优化[D]. 无锡: 江南大学, 2013.
[5] 张胜利.产反式-4-羟基-L-脯氨酸大肠杆菌菌株的改造及发酵条件初步研究[D]. 无锡: 江南大学, 2015.
[6] 许虹, 窦文芳, 许泓瑜,等.不同供氧水平对L-精氨酸分批发酵过程的影响[J]. 化工学报, 2008, 59(9):2 295-2 301.
[7] 陈晓博, 罗雪粤, 张淑荣,等. 生产L-精氨酸的脯氨酸营养缺陷型菌株的选育[J]. 北京化工大学学报(自然科学版) , 2011, 38(6): 83-86.
[8] LEE S Y, PARK J M, LEE J H, et al. Interaction of transcriptional repressor ArgR with transcriptional regulator FarR at theargBpromoter region inCorynebacteriumglutamicum[J]. Ashaipplied and Environmental Microbiology, 2011, 77(3): 711-718.
[9] SOO Y L, HWA S S , PARK J S, et al.Proline reduces the binding of transcriptional regulator ArgR to upstream ofargBinCorynebacteriumglutamicum[J]. Applied Genetics AndMolecular Biotechnology, 2010, 86(1): 235-242.
[10] 胡丹丹.无外源L-脯氨酸发酵生产反式-4-羟脯氨酸重组大肠杆菌的构建和发酵优化[D]. 无锡: 江南大学, 2015.
[11] DATSENKO K A, WANNER B L. One-step inactivation of chromosomal genes inEscherichiacoliK-12 using PCR products[J]. Proceedings of the National Academy of Sciences of the United States of America, 2000, 97(12): 6 640-6 645.
[12] POTEETE A R, FENTON A C. Genetic requirements of phage lambda red-mediated gene replacement inEscherichiacoliK-12[J]. Journal of Bacteriology, 2000, 182(8): 2 336-2 340.
[13] BABA T, ARA T, HASEGAWA M, et al. Construction ofEscherichiacoliK-12 in-frame, single-gene knockout mutants: the Keio collection[J]. Molecular Systems Biology, 2006,2(1): 1-11.
[14] DOUBLET B, DOUARD G,TARGANT H, et al. Antibiotic marker modifications of λ Red and FLP helper plasmids, pKD46 and pCP20,for inactivation of chromosomal genes using PCR products in multidrug-resistant strains[J]. Journal of Microbiological Methods, 2008, 75 (2): 359-361.
[15] SHIBASAKI T, OZAKI A, MORI H. Enzymatic production oftrans-4-hydroxy-L-proline by regio- and stereospecific hydroxylation ofL-proline[J]. Bioscience Biotechnology & Biochemistry,2000, 64(4): 746-750.
[16] 陈雪岚, 许正宏, 陶文沂. 钝齿棒杆菌产精氨酸关键酶分析[J]. 食品科学, 2005, 26(3): 35-38.
Fermentative production oftrans-4-hydroxyproline with arginine-deficient strain
WANG Xiao-jiao,ZHANG Zhen-yu*,SUN Fu-bao*,YU Lin
(College of Bioengineering, Jiangnan University, Wuxi 214122, China)
In order to obtain high-yield strains oftrans-4-hydroxyproline, based on the guidance ofE.colimetabolic network model and usingE.coliBL21 (DE3)ΔputAas the original strain, geneargBwas knocked out successfully, which blocked metabolic pathways branch ofL-glutamic acid,L-proline synthetic precursors and increased synthesis ofL-proline metabolic flux. The constructed arginine-deficient strainE.coliBL21 (DE3)ΔputAΔargBwas transformed with the expression plasmid pUC19-proB2A-Ptrp2-hyp. The expression plasmid contains the mutant geneproB2encoding glutamate kinase that significantly reducedL-proline feedback inhibition. Fermentation results indicated thatthetrans-4-hydroxyproline production of recombinant strain reached 312.67 mg/L after addition of 600 mg/L exogenous arginine, which increased by 25.29% compared withE.coliBL21(DE3)ΔputA/ pUC19-proB2A-Ptrp2-hyp.
Escherichiacoli;trans-4-hydroxyproline; knockout; proline
10.13995/j.cnki.11-1802/ts.201701005
硕士研究生(张震宇教授,孙付保副教授为通讯作者,E-mail:zhangzy@gmail.com;fubaosun@jiangnan.edu.cn)。
国家自然科学基金(30970058);江苏省自然科学基金(BK2012554);工业生物技术教育部重点实验室(江南大学)开放课题基金(KLIB-ZR200801)
2016-07-18,改回日期:2016-09-09
猜你喜欢
纺织标准与质量(2022年1期)2022-07-12
中国生殖健康(2020年5期)2021-01-18
中国生殖健康(2018年5期)2018-11-06
中国酿造(2018年7期)2018-08-10
食品与机械(2018年5期)2018-07-16
中国民族医药杂志(2016年5期)2016-05-09
食品安全导刊(2016年15期)2016-03-27
中外医疗(2015年11期)2016-01-04
中国当代医药(2015年30期)2015-03-01
中国现代医生(2014年12期)2014-06-10
