
64例胸膜恶性间皮瘤的临床病理分析
2017-01-16 11:07赵雨冯瑞娥曾瑄孟芝兰武莎菲罗玉凤曹金伶崔全才
中华肺部疾病杂志(电子版) 2016年6期
赵雨 冯瑞娥 曾瑄 孟芝兰 武莎菲 罗玉凤 曹金伶 崔全才
·论著·
64例胸膜恶性间皮瘤的临床病理分析
赵雨 冯瑞娥 曾瑄 孟芝兰 武莎菲 罗玉凤 曹金伶 崔全才
目的探讨恶性胸膜间皮瘤(malignant pleural mesothelioma, MPM)的临床病理特征及诊断与鉴别诊断。方法对64例MPM的临床病理特征、免疫表型、p16基因FISH检测结果进行分析,并复习相关文献。结果64例MPM中男性43例,女性21例,发病年龄12~78岁。标本获得方式包括手术切除(14/64,21.9%),胸腔镜下切取活检(18/64,28.1%)及粗针穿刺活检(32/64,50%)。组织学亚型包括上皮样型41例,双相型20例以及肉瘤样型3例。对其中46例MPM及另外24例间皮增生的患者进行p16基因纯合性缺失FISH检测,46例MPM中成功检测29例,发现15例存在p16基因的纯合性缺失(15/29,51.7%),而24例间皮增生中仅见1例(该例镜下间皮细胞增生十分显著,后经随访证实为间皮瘤)。对15例p16基因纯合性缺失的MPM患者进行随访,其中7例在诊断后的1年内死亡(随访1~12个月),3例存活至今(随访5~17个月),中位生存时间9个月,另5例失访。结论MPM是一种少见但发病率逐年上升的恶性肿瘤,目前的治疗手段效果欠佳,预后差。其组织学形态多样,诊断困难,应用免疫组化对诊断与鉴别诊断有很大帮助,而p16基因纯合性缺失FISH检测则对良、恶性间皮增生的鉴别有重要意义。
胸膜恶性间皮瘤; p16基因; FISH; 免疫组化; 诊断,病理
胸膜恶性间皮瘤(malignant pleural mesothelioma, MPM)是一种少见的恶性肿瘤,近年来发病率及病死率呈逐年递增的趋势,虽然已经发现了50多年,但到目前为止仍然没有理想的处理对策,预后很差。我国由于没有完全禁止石棉的生产使用,预计将来很长一段时间MPM的发病率将持续增长,正确及时的诊断及治疗对患者的预后至关重要。MPM由于组织形态多样其诊断及鉴别诊断存在困难,特别是与反应性增生间皮细胞的鉴别一直都是一个难题,很容易误诊。本研究对64例MPM的形态学特点、免疫表型和分子表型进行分析,并复习相关文献,以期增加对这种少见疾病的诊断经验,避免误诊。
资料及方法
一、一般资料
收集北京协和医院病理科1999年1月至2014年4月间诊断的MPM共64例。标本的获取方式包括手术切除、腔镜下活检及穿刺活检。并收集2012年至2014年间诊断的间皮增生病例共24例作为对照组。
二、研究方法
1. 组织学:标本采用4%甲醛固定,石蜡包埋,常规切片,HE染色,所有病例的诊断都重新经过2位正高职称病理医师的共同复核。
2. 免疫组织化学: 使用全自动Benchmark XT染色仪器Ventana( Arizona, USA), 采用EnVision染色法。抗体Calretinin、MC、CK5/6、D2-40、WT-1、Desmin、EMA、AE1/AE3、Vimentin、CEA、TTF-1、P53等均购自北京中杉金桥生物技术有限公司。
3. p16基因纯合型缺失FISH检测: 采用p16(CDKN2A)/CEP9双色探针( Abbott Molecular, Des Plaines, IL),p16(CDKN2A)显示橘红色荧光信号,CEP9(9号染色体着丝粒)显示绿色荧光信号。石蜡切片与HE切片对比后标出待杂交区域,二甲苯、100%乙醇脱蜡,酶消化及脱水处理后于杂交组织中央加10ul探针,上自动杂交仪(ThermoBrite Elite system, Leica)变性、杂交过夜,经洗片、DAPI复染后封片。
三、结果判定
1. 免疫组化: AE1/AE3、Vimentin、Calretinin、CK5/6、Desmin、CEA阳性为胞质着色,MC(Mesothelial cell)阳性为胞膜着色,EMA、D2-40阳性为膜/质着色,P53、TTF-1、WT-1阳性为胞核着色,所有肿瘤细胞均不着色为(-);个别细胞着色为(±);5%~25%为(+),26%~50%为(++),>50%为(+++)。
2. p16基因纯合型缺失FISH检测: 在FISH处理切片上针对事先确定的MPM肿瘤区域和对照组表面间皮增生区域进行判读,每一例至少计数60个完整细胞,尽量排除重叠细胞,≥10%的病变细胞核橘红色信号纯合丢失并至少有一个绿色信号判读为阳性。
结 果
一、一般资料
64例MPM患者中男性43例(67%),女性21例(33%),男女比2.05︰1,发病年龄从12岁~78岁,中位年龄56.5岁。对15例p16基因纯合性缺失的病例进行随访,其中7例在诊断后的1年内死亡(随访1~12个月),3例存活至今(随访5~17个月),中位生存时间9个月,另5例失访。
二、病理检查
1. 大体检查: 64例MPM患者的标本获得方式包括:粗针穿刺活检32例(32/64,50%),穿刺组织灰白灰粉,每例2~4条,直径0.05 cm,长度0.5~1 cm;胸腔镜下切取活检18例(18/64,28.1%),组织数量及大小不等,一般为灰白、灰粉或灰褐色,部分呈囊皮样,一侧光滑,一侧粗糙。手术切除标本14例(14/64,21.9%),大部分为囊皮样组织,大小从2.5 cm×1.5 cm×0.1 cm~7.5 cm×5 cm×2 cm,一侧面光滑,一侧面粗糙附暗褐色坏死物。
2. 镜下检查: 64例MPM患者的组织学分型情况详见表1。其中上皮样型41例(41/64,64%),包括了管状乳头型、腺泡型、小梁型、实性型、多形性以及2例罕见的小细胞型。管状乳头型由腺管状结构和具有结缔组织轴心的乳头状结构混合而成,被覆上皮呈多角形、立方形或扁平状,细胞异型性大多不明显,见图1。腺泡型由被覆立方上皮的腺体所构成,腺体结构常见分支或被拉长成裂隙状,见图2。小梁型MPM由形态温和的多角形细胞排列成小梁状。实性型则由片状或巢团状排列的肿瘤细胞组成,细胞圆形、卵圆形或立方形,胞浆丰富、嗜酸,核圆形,常可见核仁,异型性多不明显,细胞之间连接不紧密,见图3。而多形性MPM的肿瘤细胞异型性明显,细胞间的连接相对其他亚型更为松散,核大且畸形、核仁明显,常见核分裂像,见图4。另有2例罕见的小细胞型MPM,其中1例表现为形态温和一致的小圆细胞呈实性片状、小梁状排列,胞浆少,核浆比高,见图5。另外1例则相对异型性更大,核浆比很高,几乎见不到胞浆。本组病例共有肉瘤样型3例,由梭形细胞呈弥漫或束状分布,1例细胞形态温和,1例促纤维增生型可见间质的增生及硬化,1例局部可见明显细胞异型并伴有肿瘤性坏死。双相型共有20例,表现为上皮样和肉瘤样两种结构混合存在,部分病例可见两种结构之间的形态移行,见图6。其中1例双相型MPM为上皮样型合并淋巴组织细胞样型,见图7,及经典肉瘤样型,伴明显细胞异型性、核分裂及肿瘤性坏死,可见淋巴组织细胞样型和上皮样型间的形态学过渡。

表1 64例MPM的组织学亚型
注:MPM:恶性胸膜间皮瘤
3. 免疫组化: 64例MPM的免疫组化结果,见表2。
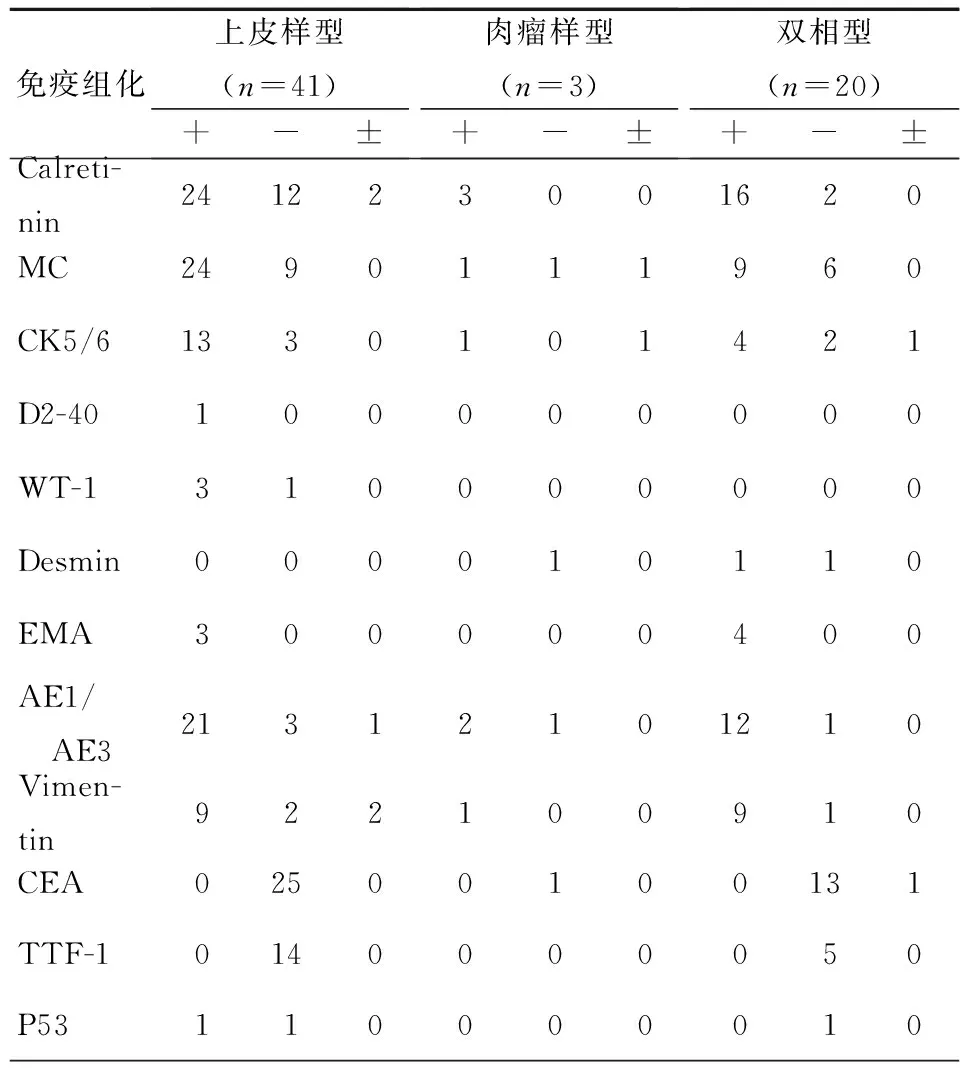
表2 64例MPM的免疫组化结果
注:MPM:恶性胸膜间皮瘤;Calretinin:钙网膜蛋白;MC:间皮细胞;Desmin:结蛋白;EMA:上皮膜抗原;Vimentin:波形蛋白;CEA:癌胚抗原;TTF-1:甲状腺转录因子-1
4. p16基因纯合性缺失FISH检测: 对其中46例MPM及另外24例间皮增生的病例进行p16基因纯合性缺失FISH检测。46例MPM中成功检测29例,其中15例存在p16基因的纯合性缺失(15/29,51.7%),见表3,见图8;24例间皮增生中见有1例存在该基因的纯合性缺失。

表3 15例p16基因纯合性缺失病例的详细情况
注:NA:失访; mo:月
讨 论
一、流行病学
恶性胸膜间皮瘤是一种少见的恶性肿瘤,近年来发病率不断上升,在一些国家和地区已经不再少见[1]。其首要致病因素是接触石棉,主要包括以下6种可以形成极细纤维的石棉:温石棉、青石棉、铁石棉、直闪石、透闪石和阳起石。这些纤细的石棉纤维可被深入的吸到肺里并穿破组织直达胸膜,其可能通过长期刺激间皮细胞导致不断修复增生、干扰有丝分裂导致细胞非整倍体以及产生毒性氧自由基和干扰细胞信号传导通路等途径致使肿瘤发生[2]。虽然多数欧美国家已经禁止使用石棉,但从石棉暴露到发生MPM的潜伏期很长(一般可达20~50年),预计这些国家和地区在未来10年中MPM的发病率将继续增加并达到顶峰[3]。我国虽然禁止使用闪石类石棉,但仍在开采使用温石棉,所以在未来的很长一段时间内,我国MPM的发病率将持续增长。

图1 管状乳头型MPM,可见腺管状结构和具有纤维血管轴心的乳头(HE×10);图2 腺泡型MPM,纤维间质中可见挤压成角的腺体(HE×10);图3 实性型MPM,形态温和的肿瘤细胞呈巢片状分布,细胞间连接不紧密,可见裂隙(HE×10);图4 多形性MPM,细胞异型性明显,核大畸形、核仁明显,连接更为松散(HE×20);图5 小细胞型MPM,形态温和一致的小圆细胞呈实性片状、小梁状排列,胞浆少,核浆比高(HE×10);图6 双相型MPM,上皮样和肉瘤样两种结构混合存在,该区域可见两种结构之间的形态移行(HE×40);图7 一例双相型MPM中的淋巴组织细胞样型区域,细胞散在,异型性明显,胞浆丰富类似组织细胞,背景中见大量淋巴细胞及浆细胞(HE×40);图8 p16基因纯合型缺失FISH检测阳性,荧光显微镜下见该视野内多数细胞只有绿色信号(9号染色体着丝粒)而没有橘红色信号(p16基因所在位置)
二、临床表现
MPM典型的临床表现为劳力性呼吸困难、持续性胸壁疼痛以及胸水,全身症状如体重下降和疲劳一般出现在病程后期,一旦出现多提示预后不良[4-5]。以上症状并不为MPM所特有,其诊断还需要结合影像学以及病理检查。MPM患者的X线检查多表现为大量胸水,但因此胸膜病变往往显示不清,CT和MRI可以更好的判断肿瘤范围,多表现为胸膜增厚、胸膜斑块或局部肿块,肿瘤可以沿着脏器表面或裂隙呈环状浸润[6]。
三、组织学分型
间皮瘤按组织学形态分为上皮样、肉瘤样和双相型3个亚型,其中最常见的是上皮样型,该亚型在我们的病例中占到了64%。上皮样型中最常见的是管状乳头型、腺泡型以及实性型,其他少见的亚型包括蜕膜样、透明细胞、印戒样、腺样囊性型、横纹肌样、多形性、嗜酸性、肾小球样以及小细胞型等,但目前其中一些少见亚型是否真的存在仍存有争议[7]。本研究中虽有以上一些亚型单独存在的病例,但大多数都含有两种及以上形态(18/41,44%),而且本组病例以粗针穿刺为主(32/64,50%),在这些有限的标本中不可能明确肿瘤所有的成分,所以实际上混合多种形态的病例可能会更多。而肉瘤样型则分为经典型(梭形细胞)、促纤维增生型、异源分化型(如骨肉瘤、软骨肉瘤等)以及淋巴组织细胞样型(该型也可划为上皮样型)[8]。
四、诊断
MPM的病理诊断主要包括细胞学和组织学两方面:①细胞学检查:MPM最常见的细胞学标本是胸水,一般在胸水中只能看到上皮样型的肿瘤细胞。涂片中细胞量丰富,肿瘤细胞排列成大的桑葚样球团,细胞大、核异性明显、细胞间有时可见“开窗”现象。而肉瘤样型MPM的细胞很难脱落到胸水中,加上一些MPM并不伴有胸水,此时的细胞学诊断需要细针穿刺。由于细胞学取材有限以及无法观察肿瘤是否有浸润等原因,其在明确诊断MPM时存在先天不足,虽然目前的观点存在争议,但可以明确的是细胞学可以在临床证据充分的情况下提出疑似诊断并为下一步的组织学检查指明方向[9];②组织学诊断:因为胸腔镜可以全面检查胸膜的情况并能取到充分的组织以供病理诊断,目前欧美的指南均一致推荐其作为MPM的首选诊断方法[3,10]。MPM的组织学形态变化多样,上皮样型MPM的细胞可以从形态变异型性较少到明显异型,可以排列成腺样、乳头状、小梁状、实性等结构。而肉瘤样型则可以由束状或杂乱排列梭形细胞到显著间变以及奇异的多核瘤巨细胞所构成,加上可以出现异源性的肿瘤成分以及一些罕见亚型的特殊形态。MPM的诊断经常很困难,确诊需要结合免疫组化、分子生物学、临床以及影像学综合考虑。
五、鉴别诊断
由于形态多样,MPM需要鉴别的疾病很多,总的来说上皮样型MPM需要和癌以及其他的上皮样肿瘤鉴别,肉瘤样型需要和肉瘤以及其他一些梭形细胞肿瘤相鉴别,而双相型则需要鉴别其他具有双相形态的肿瘤,如滑膜肉瘤、癌肉瘤等[8]。由于单纯依靠形态学区分极其困难,目前MPM与其他肿瘤的鉴别诊断主要依靠多种免疫组化抗体的搭配使用。间皮瘤可以选择的阳性标记包括Calretinin、WT-1、D2-40、CK5/6、MC等,另据我们的经验AE1/AE3和Vimentin搭配使用效果也很好。以下是国际间皮瘤学会2012年指南中提到的一些MPM的鉴别诊断以及推荐抗体组合[8]:①肺腺癌,表达TTF-1、Napsin A、CEA以及上皮性肿瘤的通用标记Ber-EP4、MOC-31等,MPM这些标记均为阴性而间皮标记阳性,容易鉴别;②肺鳞癌,最特异的抗体为p63或p40,几乎所有的肺鳞癌都有弥漫的核强阳性表达,而MPM只有极少一部分弱阳性表达,因为Calretinin、D2-40以及CK5/6在两者都有不同程度的表达,所以最好选择WT-1作为间皮的标记;③肺小细胞癌,可以累及胸膜并表现为类似间皮瘤,组织学形态上也与小细胞型MPM有很多相似之处,如细胞密集、高核浆比等。有作者总结小细胞型MPM相比小细胞肺癌胞浆更多、核呈空泡状、染色质更匀细并往往有小核仁、没有凋亡小体,核分裂像也远低于小细胞肺癌,而且小细胞型MPM不表达TTF-1、MOC-31以及神经内分泌标记可与小细胞肺癌进一步鉴别[7]。需要注意的是Calretinin在很大一部分肺小细胞癌中也有表达,单纯靠它不能鉴别两者[11];④乳腺癌,表达ER、PR、GCDFP-15、Mammaglobin,依靠这些抗体可以与MPM鉴别;⑤恶性黑色素瘤,形态多样,主要由上皮样细胞和梭形细胞构成且可以没有色素,有时容易和MPM混淆,S-100、HMB-45和Melan-A有助于鉴别。需要注意的是转移性恶性黑色素瘤有时可以发生表型去分化,即失去以上抗体的表达,此时的诊断更需谨慎,最好明确之前有无恶性黑色素瘤的病史,如能行电镜观察有无黑色素小体是最为稳妥的鉴别办法[12];⑥上皮样血管内皮瘤,细胞呈上皮样,排列成巢状或条索状,异型性一般不明显,一小部分病例可原发于胸膜,如果对其不熟悉容易误诊为间皮瘤,CD31和CD34对鉴别很有帮助;⑦血管肉瘤,很少见发生于胸膜,不过其形态学变化很大,可以从梭形到上皮样不等,容易和MPM混淆,鉴别也主要依靠CD31和CD34;⑧滑膜肉瘤,可以导致胸膜弥漫增厚,其双相分化的形态需要和双相型MPM鉴别。滑膜肉瘤的梭形细胞分布密集、胞浆稀少,至少局部能看到血管周细胞瘤样结构,加上CD99、Bcl-2阳性可以和MPM鉴别。在一些鉴别困难的病例做t(X;18)(p11;q11)染色体易位检测则能很好的帮助明确滑膜肉瘤的诊断;⑨淋巴瘤,上皮样型MPM有时会呈实性、单一的、相对不粘附的细胞巢团,需要和淋巴瘤鉴别,大多数情况下CD3、CD20加上间皮的标记足以区分两者;⑩纤维肉瘤、癌肉瘤,两者均需要和肉瘤样型MPM鉴别,纤维肉瘤上皮标记阴性,加上Calretinin和D2-40可以区分两者;而癌肉瘤上皮标记为阳性,MPM与之鉴别需要明确的Calretinin和D2-40阳性,而WT-1和CK5/6此时对鉴别诊断帮助不大。
由于没有特异的标记,MPM与反应性间皮增生的鉴别一直是一个难题,尤其是在细胞学诊断时更为困难。有作者曾提出用Desmin和EMA的抗体组合来区分两者,但特异性只有80%左右[13]。 近来用FISH检测p16基因纯合性缺失在鉴别恶性间皮瘤与反应性间皮增生之中的作用被越来越多的学者所认可,其报道的敏感性约为59%~79%,而特异性均为100%[14-16]。本组病例也只在MPM病例组中检测出了该基因的纯合性缺失,而对照组中唯一的1例阳性病例镜下虽然没有浸润但间皮细胞增生十分显著,后经随访证实为恶性间皮瘤,患者于诊断后的12个月内死亡。于此相应的,有作者认为在表面间皮细胞增生的病例中如果FISH检测到了p16基因的纯合性缺失,即便是没有明确的浸润,在临床证据充分的前提下也可以做出恶性间皮瘤的诊断[17]。
六、细胞遗传学改变
传统的细胞遗传学发现大多数MPM都存在不同程度的染色体异常,包括非整倍体及染色体结构改变等。近来随着二代测序等检测手段的发展,进一步发现MPM存在一些关键基因的改变,如p16/CDKN2A、BAP1、NF2以及CUL1,其中最为常见的是位于9p21的p16/CDKN2A基因的纯合性缺失,文献报道其发生率最高可以达到80%[8,18]。除了纯合性缺失,p16/CDKN2A基因在MPM中还可以发生点突变及DNA甲基化,但相对较少而且可以在良性间皮细胞中检测到,所以在鉴别良恶性时应用价值不大[19]。另外需要注意的是p16/CDKN2A基因的改变虽在MPM中发生率很高但并不为其所特有,不能靠它来帮助MPM与其他恶性肿瘤的鉴别诊断[8]。
七、预后判断
目前MPM患者的体力状况评分和组织学分型是唯一具有临床意义的预后因素[3]。总的来说,上皮样MPM的预后好于肉瘤样型以及双相型。但近来发现一些例外,如多形性MPM的生物学行为更具有侵袭性,预后类似于肉瘤样型以及双相型,而淋巴组织细胞样型的预后则更类似于上皮样型,有作者认为以上二者的类型应该重新划分[8, 20-21]。另外,一直以来MPM的组织学分级与预后的关系没有被提及,但最近的研究指出上皮样型MPM中核分裂像以及核异型性与预后密切相关[22]。而p16基因的纯合性缺失也被多个研究证实是一种提示预后不良的因素[23-24]。
八、治疗
MPM的治疗一直很困难,目前的手段主要包括外科手术、化疗以及放疗。外科手术有胸膜外全肺切除术和减瘤胸膜切除术/胸膜剥脱术,其中后者虽然达不到治愈目的但可以明显缓解症状,而且可在胸腔镜下完成,对患者的损伤较小。对MPM患者能否从化疗中获益目前还有争议,欧洲呼吸学会/欧洲胸外科医师学会的诊疗指南中指出尽管顺铂单药不能作为标准治疗方案,但顺铂联合培美曲塞或者雷替曲塞在缓解率和生存期方面均优于顺铂单药。同样2013年美国国立综合癌症网络(NCCN)胸膜恶性间皮瘤指南中推荐的一线化疗药物也以培美曲塞联合铂类为主[3]。没有预后资料表明激进的半胸放疗可以改善MPM患者的预后,所以目前放疗仅仅作为一种辅助治疗手段或者用来缓解症状[25]。另外,目前已在进行试验的生物靶向药物均没有观察到可靠的疗效[3]。所以,就目前来看MPM的治疗可能还有很长的一段路需要走。
1 Tsiouris A, Walesby RK. Malignant pleural mesothelioma: current concepts in treatment[J]. Nat Clin Pract Oncol, 2007, 4(6): 344-352.
2 Robinson BWS, Musk AW, Lake RA. Malignant mesothelioma[J]. Lancet, 2005, 366(9483): 397-408.
3 Scherpereel A, Astoul P, Baas P, et al. Guidelines of the European Respiratory Society and the European Society of Thoracic Surgeons for the management of malignant pleural mesothelioma[J]. Eur Respir J, 2010, 35(3): 479-495.
4 Tsiouris A, Gourgoulianis KI, Walesby RK. Current trends in the management of malignant pleural mesothelioma[J]. Expert Rev Anticancer Ther, 2006, 6(6): 831-833.
5 Scott B MS, Lake R, Robinson BWS. Malignant mesothelioma. In: Hanson H e, editor. Textbook of lung cancer[M]. London: Martin Dunitz, 2000: 273-293.
6 Travis WD, Brambilla E, Müller-Hermelink H K, et al. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. World Health Organization classification of tumours[M]. Lyon: IARC Press, 2004: 130.
8 Husain AN, Colby T, Ordonez N, et al. Guidelines for pathologic diagnosis of malignant mesothelioma: 2012 update of the consensus statement from the International Mesothelioma Interest Group[J]. Arch Pathol Lab Med, 2013, 137(5): 647-667.
9 Sheaff M. Should cytology be an acceptable means of diagnosing malignant mesothelioma?[J]. Cytopathology, 2011, 22(1): 3-4.
10 Ettinger DS, Akerley W, Borghaei H, et al. Malignant pleural mesothelioma[J]. J Natl Compr Cancer Netw, 2012, 10(1): 26-41.
11 Lugli A, Forster Y, Haas P, et al. Calretinin expression in human normal and neoplastic tissues: a tissue microarray analysis on 5233 tissue samples[J]. Hum Pathol, 2003, 34(10): 994-1000.
12 刘彤华. 诊断病理学(第3版)[M]. 北京: 人民卫生出版社, 2013: 1106-1108.
13 Attanoos RL, Griffin A, Gibbs AR. The use of immunohistochemistry in distinguishing reactive from neoplastic mesothelium. A novel use for desmin and comparative evaluation with epithelial membrane antigen, p53, platelet-derived growth factor-receptor, P-glycoprotein and Bcl-2[J]. Histopathology, 2003, 43(3): 231-238.
14 Chiosea S, Krasinskas A, Cagle PT, et al. Diagnostic importance of 9p21 homozygous deletion in malignant mesotheliomas[J]. Mod Pathol, 2008, 21(6): 742-747.
15 Savic S, Franco N, Grilli B, et al. Fluorescence in situ hybridization in the definitive diagnosis of malignant mesothelioma in effusion cytology[J]. Chest, 2010, 138(1): 137-144.
16 Monaco SE, Shuai Y, Bansal M, et al. The diagnostic utility of p16 FISH and GLUT-1 immunohistochemical analysis in mesothelial proliferations[J]. Am J Clin Pathol, 2011, 135(4): 619-627.
17 Hwang H, Tse C, Rodriguez S, et al. p16 FISH deletion in surface epithelial mesothelial proliferations is predictive of underlying invasive mesothelioma[J]. Am J Surg Pathol, 2014, 38(5): 681-688.
18 Guo G, Chmielecki J, Goparaju C, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma[J]. Cancer Res, 2015, 75(2): 264-269.
19 Pu RT, Sheng ZM, Michael CW, et al. Methylation profiling of mesothelioma using real-time methylation-specific PCR: a pilot study[J]. Diagn Cytopathol, 2007, 35(8): 498-502.
20 Ak G, Metintas S, Metintas M, et al. Prognostic factors according to the treatment schedule in malignant pleural mesothelioma[J]. J Thorac Oncol, 2009, 4(11): 1425-1430.
21 Kadota K, Suzuki K, Sima CS, et al. Pleomorphic epithelioid diffuse malignant pleural mesothelioma: a clinicopathological review and conceptual proposal to reclassify as biphasic or sarcomatoid mesothelioma[J]. J Thorac Oncol, 2011, 6(5): 896-904.
22 Kadota K, Suzuki K, Colovos C, et al. A nuclear grading system is a strong predictor of survival in epitheloid diffuse malignant pleural mesothelioma[J]. Mod Pathol, 2012, 25(2): 260-271.
23 Lopez-Rios F, Chuai S, Flores R, et al. Global gene expression profiling of pleural mesotheliomas: overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction[J]. Cancer Res, 2006, 66(6): 2970-2979.
24 Dacic S, Kothmaier H, Land S, et al. Prognostic significance of p16/cdkn2a loss in pleural malignant mesotheliomas[J]. Virchows Arch, 2008, 453(6): 627-635.
25 Porpodis K, Zarogoulidis P, Boutsikou E, et al. Malignant pleural mesothelioma: current and future perspectives[J]. J Thorac Dis, 2013, 5 (Suppl 4): S397-S406.
(本文编辑:黄红稷)
赵雨,冯瑞娥,曾瑄,等. 64例胸膜恶性间皮瘤的临床病理分析[J/CD]. 中华肺部疾病杂志: 电子版, 2016, 9(6): 590-595.
Malignant pleural mesothelioma: clinicopathologic analysis in a consecutive series of 64 patients from a single institution in China
ZhaoYu,FengRuie,ZengXuan,MengZhilang,WuShafei,LuoYufeng,CaoJinling,CuiQuancai.
DepartmentofPathology,PekingUnionMedicalCollegeHospital,ChineseAcademyofMedicalSciences&PekingUnionMedicalCollege,Beijing, 100730,China
CuiQuancai,Email:cuiqc@sina.com
Objective To investigate the clinicopathologic characteristics, pathologic diagnosis and differential diagnosis of malignant pleural mesothelioma (MPM). Methods The clinical and histological findings, immunophenotype, p16 FISH deletion and prognosis of 64 cases of MPMs were evaluated with review of the relevant literature. Results All of 43 patients were male and 21 were female, with age from 12 to 78 years. Specimens from surgical removal(14/64,21.9%),biopsy by thoracscope(18/64,28.1%),coarse needle biopsy(32/64, 50%). There were 41 epithelioid MPMs, 20 biphasic MPMs and 3 sarcomatoid MPMs. p16 FISH was used to investigate 29 MPMs and 24 surface mesothelial proliferation cases successfully, homozygous deletion of p16 was find in 15/29 MPMs and 1/24 mesothelial proliferation cases. Of the 15 patients with p16 homozygous deletion, 7 died in 1 year (followed 1-12 months) and 3 survived till now (followed 5-17 months), the median survival was 9 months, the other 5 patients lost to follow-up. Conclusions MPM is a rare disease but its incidence is increasing worldwide, there are no efficient cures for this disease by now. It is difficult to diagnose MPM because of its morphological variety, immunohistochemistry could be helpful and p16 deletion FISH is becoming more important in the differentiation between surface mesothelial proliferation and MPM.
Malignant pleural mesothelioma; p16 gene; FISH; Immunohistochemistry; Diagnosis
10.3877/cma.j.issn.1674-6902.2016.06.002
国家自然科学基金委青年科学基金项目(81602162)
100730 北京,中国医学科学院 北京协和医学院 北京协和医院病理科
崔全才,Email:cuiqc@sina.com
R734.2
A
2016-10-12)
猜你喜欢
数学物理学报(2022年5期)2022-10-09
山东冶金(2022年2期)2022-08-08
临床肺科杂志(2022年8期)2022-07-28
中国临床医学影像杂志(2022年2期)2022-05-25
中国药学药品知识仓库(2022年7期)2022-05-10
现代临床医学(2021年4期)2021-07-31
粉末冶金技术(2021年3期)2021-07-28
云南医药(2021年3期)2021-07-21
中国现代医药杂志(2020年10期)2020-12-14
中国临床医学影像杂志(2019年4期)2019-06-18
