
基于生物信息学方法的荞麦属microRNAs及其靶基因预测
2017-01-05 06:13高帆宋韡谢树莲
山西农业科学 2016年9期
高帆,宋韡,谢树莲
(1.山西大学生命科学学院,山西太原030006;2.山西体育职业学院,山西太原030006)
基于生物信息学方法的荞麦属microRNAs及其靶基因预测
高帆1,宋韡2,谢树莲1
(1.山西大学生命科学学院,山西太原030006;2.山西体育职业学院,山西太原030006)
MicroRNA是一类具有转录后基因调控功能的非编码小分子RNA。以传统的试验方法发现和鉴定新的miRNA是一项繁琐的工作。以生物信息学方法从数据库中筛选miRNAs及其靶基因能够极大地提高效率。植物中已报道了大量的miRNAs,但荞麦属的尚未见报道。通过荞麦ESTs和GSSs的严格筛选,共预测到13个荞麦属miRNAs(分属11个miRNA家族)。预测的miRNAs在不同的植物中表现出保守性,miRNA家族成员表现出多样性。共预测到17个潜在的靶基因,这些靶基因的功能包括代谢、生长发育、胁迫响应、信号转导等,表明miRNAs在植物生命活动中具有重要作用。miRNA家族系统进化分析表明,金荞麦的miRNAs进化与甜荞麦关系密切。以生物信息学方法从已知的数据库中预测并挖掘荞麦属新的miRNAs及其靶基因是可行的。
MicroRNA(miRNA);生物信息学;荞麦属;靶基因
荞麦属(Fagopyrum)是蓼科(Polygonaceae)植物中被人们熟知的类群,其中,有些种是常见杂粮作物,如甜荞麦(F.esculentum)和苦荞麦(F.tataricum)在许多国家都有栽培[1];有的种具有药用价值[2],如苦荞麦、金荞麦(F.cymosum)和其他一些野生种的提取物具有降低血糖和血脂[3-5]、抗菌、抗氧化等功效[6-7]。发展和提高作物产量、有效改善作物品质、挖掘并探究某些作物特殊的药用成分及其作用机制是当前作物研究的热点[8-9]。预测并鉴定荞麦microRNAs(miRNAs)及其靶基因,探究靶基因转录后表达调控机制将有助于进一步了解荞麦生长发育、营养代谢、药用机制以及遗传进化的详细进程。
miRNA是一类具有调控功能的非编码小分子RNA。Lee等[10]在秀丽隐杆线虫(Caenorhabditis elegans)中首次发现了这类特殊的RNA分子。大多数miRNAs长度约为19~25 nt[11]。RNA聚合酶II和III在miRNA基因转录中起着重要作用[12]。研究显示,miRNAs在植物生长发育、生物代谢、胁迫响应、信号转导及其他生命活动中均发挥着重要的作用[13]。自2002年以来,植物miRNA的研究有了快速地发展[14]。截止2016年8月,miRBase数据库已正式公布了33种植物共约8 450条miRNAs[15]。尽管如此,作为一种重要的小杂粮作物,荞麦的miRNAs还未有任何报道。预测荞麦属miRNAs将扩展植物miRNAs信息,并有助于深入研究该作物生命活动过程中的某些基因的表达调控机制。这将为定向改良荞麦种质,详细了解其药用机理,高效、精准、充分地利用现有荞麦资源,提高我国荞麦品质奠定基础。
目前,获得miRNAs的主要方法有直接克隆[16]、正向遗传[17]、深度测序[18-19]和生物信息[20]。由于材料获取和试验成本等方面的困难,生物信息学方法以其简单、快速、高效而倍受研究者的青睐[21-22]。与动物相比,一些植物的miRNA序列相对保守[23],因此,通过生物信息学方法预测荞麦属miRNAs是可行的[24]。
本研究利用荞麦的ESTs和GSSs数据库,以已知植物miRNAs为探针,采用多种生物信息手段筛选候选序列并预测其前体结构,最终确定了荞麦的miRNAs,此外还预测了miRANs的靶基因及其功能,最后分析了荞麦miRNAs在植物中的保守性、多样性及其不同家族间的系统进化关系。
1 材料和方法
1.1 序列获得
从miRBase21.0数据库中(http://www.mirbase. org/)下载所有植物的miRNAs及其前体序列,去除冗余序列,保留的序列在同一miRNAs家族中不重复并按照不同种类分组。由于荞麦的全基因组测序尚未完成,本研究以ESTs(273条)和GSSs(85条)为模板序列(从Genbank中下载获得,http://www. ncbi.nlm.nih.gov),以整理的植物miRNAs数据库为搜索序列,通过多重序列比对获取同源性较高的序列作为荞麦候选miRNA序列。
1.2 潜在miRNA序列的预测
通过考察候选miRNA序列,并将其与Rfam(http://rfam.sanger.ac.uk)及Genbank数据库比对,从中可获得潜在的荞麦miRNAs[25]。根据以下原则筛选miRNA序列:(1)不一致的碱基不超过3个;(2)百分比不超过(L-4+m)/L,其中,L是长度(碱基数),m是不一致的碱基数;(3)去除tRNA和rRNA及蛋白编码序列[26-27];(4)去除重复序列。
1.3 前体miRNA次级结构的预测
为了进一步确认潜在的miRNA序列为荞麦miRNAs,同时考察其前体是否能形成典型的颈环状二级结构,是否含有星状miRNA(miRNA*),本研究对荞麦miRNA的前体进行了预测。根据有关文献报道,植物中前体miRNA长度通常约为144.57±56.91 nt[28],本研究以200 nt miRNAs上游和下游序列包括其本身作为源序列,分析预测前体序列的二级结构[29],长度限于60~250 nt。参数设置为:(1)选择概率值为0.5;(2)扫描窗口值为5个碱基对、延伸窗口值为3个碱基对;(3)所需的前体miRNA构建次级结构,其中,miRNA和miRNA*必需位于“发夹”结构的一个臂上;(4)在臂上没有更大或更多的隆起结构;(5)最小折叠自由能不超过-75.3 kJ/mol;(6)(A+U)占比在30%~70%;(7)不一致的碱基数目不超过6,在miRNA和其互补链中无环状结构或间断出现。
1.4 靶基因的预测
用WMD3在线分析软件(http://wmd3.weigelworld.org/)结合BLASTX及Pfam软件(http://pfam. sanger.ac.uk/)预测并分析miRNAs靶基因及其功能[25-27],预测原则为:(1)不一致的碱基数不超过5个;(2)在位点2~12之间不一致的碱基数不超过1个,其中在位点10~11之间不允许有碱基错配;(3)在位点13~21之间错配的碱基数不超过4个,且不能有2个连续的错配碱基;(4)选择的结构具有最低的MFE值和最高的最小折叠自由能指数(MFEI)[30]。以WMD3中主要植物的mRNAs作为模板序列,包括模式植物和主要作物,如拟南芥、水稻和玉米。由于WMD3和Pfam中尚未公布荞麦mRNAs信息,本研究以ESTs和Genbank中的mRNAs作为比对数据。
1.5 miRNA的保守性、多样性及其家族成员间的系统进化分析
通过BLASTn比对分析不同植物中miRNAs的保守性[31-32],进而分析不同种中miRNA家族成员的数目及其保守性和多样性。在此基础上,利用MEGA3.1工具,以邻接法构建不同荞麦miRNAs家族成员间的系统进化树[33]。
2 结果与分析
2.1 miRNAs及其前体序列预测与特征分析
本研究从数据库中下载获得了33种植物的miRNAs,通过序列比对在荞麦中预测到13个新的miRNAs,筛选率仅为1.1%(表1)。
从表2可以看出,这13个预测的miRNAs的长度为17~23 nt,符合植物miRNA长度的标准[34]。有文献报道,miRNA中(A+U)比例高于(G+C),但也不绝对[35]。本研究结果中(A+U)在35.30%~73.68 %,基本与文献报道一致。
从图1可以看出,本研究获得的所有前体具有典型的“发夹”结构,miRNAs总是位于前体结构的一个臂上,以5′端居多。前体miRNAs长度为66~153 nt,平均106.77 nt,大于动物(70~80 nt)的长度[30]。研究表明,miRNAs的MFEI应不小于0.85,本研究结果在1.22~3.84,因此,预测结果是基本可信的。13个预测的miRNAs隶属11个miRNA家族,fes-miR162和fcy-miR1298家族各有2个,其余家族各有1个。
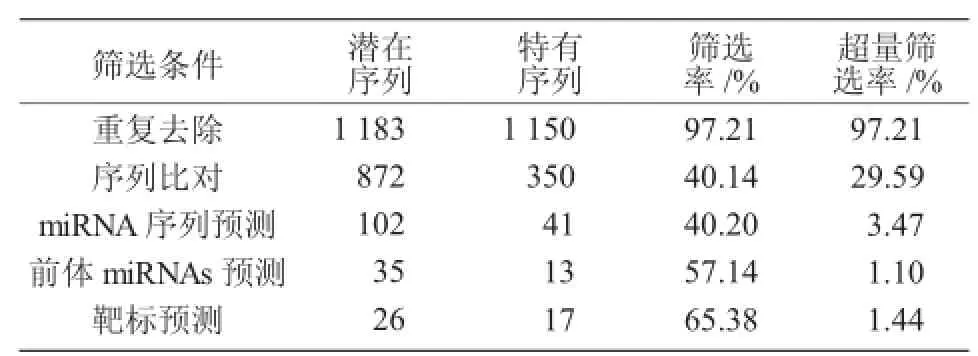
表1 潜在和特有的小分子RNAs汇总
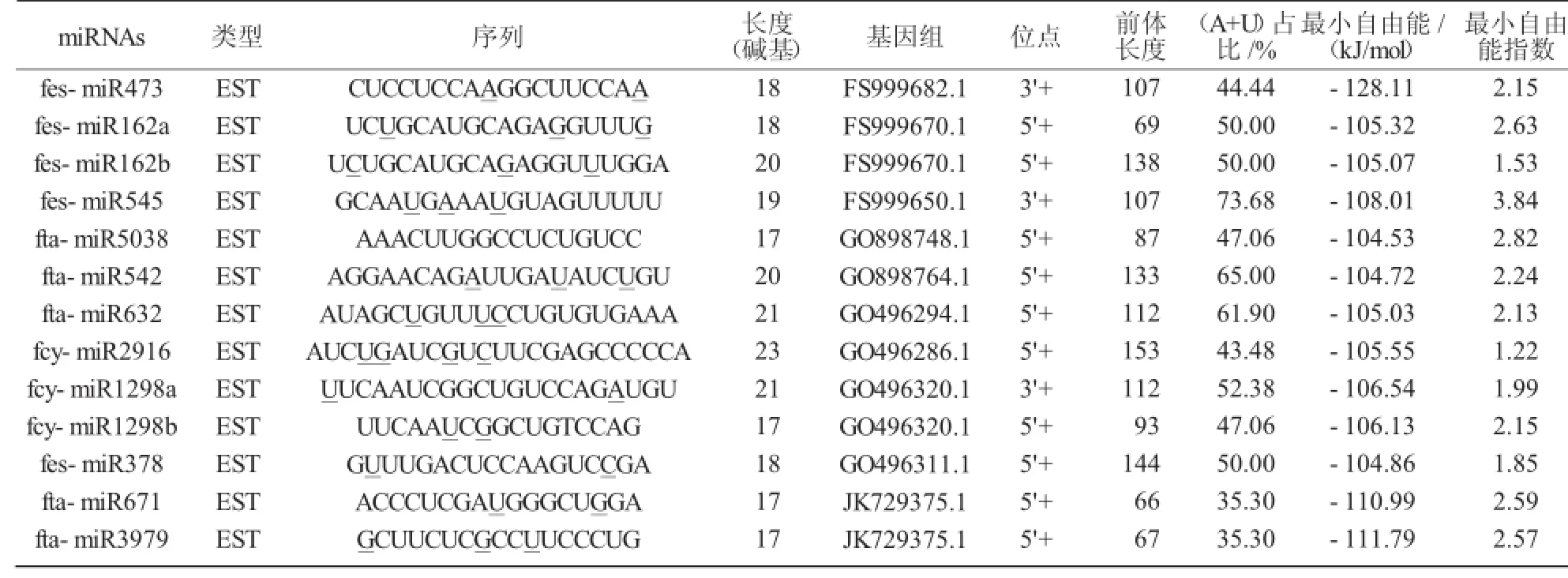
表2 预测的荞麦中miRNAs及特征

2.2miRNAs的保守性和多样性预测
本研究采用序列比对的方法分析了荞麦miRNAs的保守性和多样性。结果表明,大多数miRNAs家族是保守的,但miR162家族例外(图2)。miR473和miR1298家族在5′端的前4个碱基完全相同,与有关文献报道一致[36]。其他miRNAs也显示出与水稻、拟南芥、玉米、葡萄、毛果杨和小立碗藓的同源性。不同植物miRNAs的保守性也在一定程度上反映出其进化的保守性。
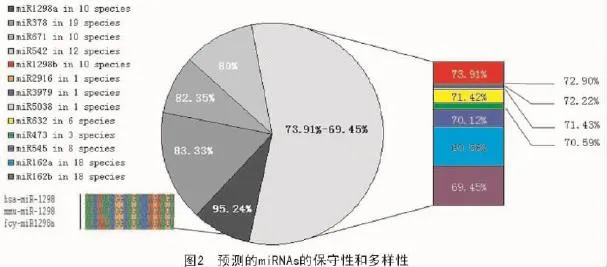
从图2还可以看出,fcy-miR1298a与现代人的hsa-miR-1298以及小鼠的mmu-miR-1298高度相似,仅在第15个位点有1个碱基的差异。在三者中发现的miR1298具有如此高的相似率是值得注意的,通常不同物种间miRNAs的保守现象只见于同界的不同种类中[37-38],并且其调控的靶基因产物往往相似。然而,动物和植物间miRNAs的保守性通常不高,且其基因产物往往不同。由于这3个物种的miR1298靶基因均是未知蛋白,并且三者的前体miRNA序列和结构完全不同。因此,不能单纯地从miR1298成熟体的保守性来推测其在人类和小鼠中也完全是保守的。
本研究发现荞麦中预测的13个miRNAs也存在于其他植物中(表3),miR378,miR542,miR671和miR1298家族至少存在于10种植物中。同时,本研究也发现一些罕见的miRNAs,如miR3979,miR5038和miR2916,仅见于1种植物中,基本属于物种特异miRNAs(novel miRNAs),这也反映了植物miRNAs的多样性。
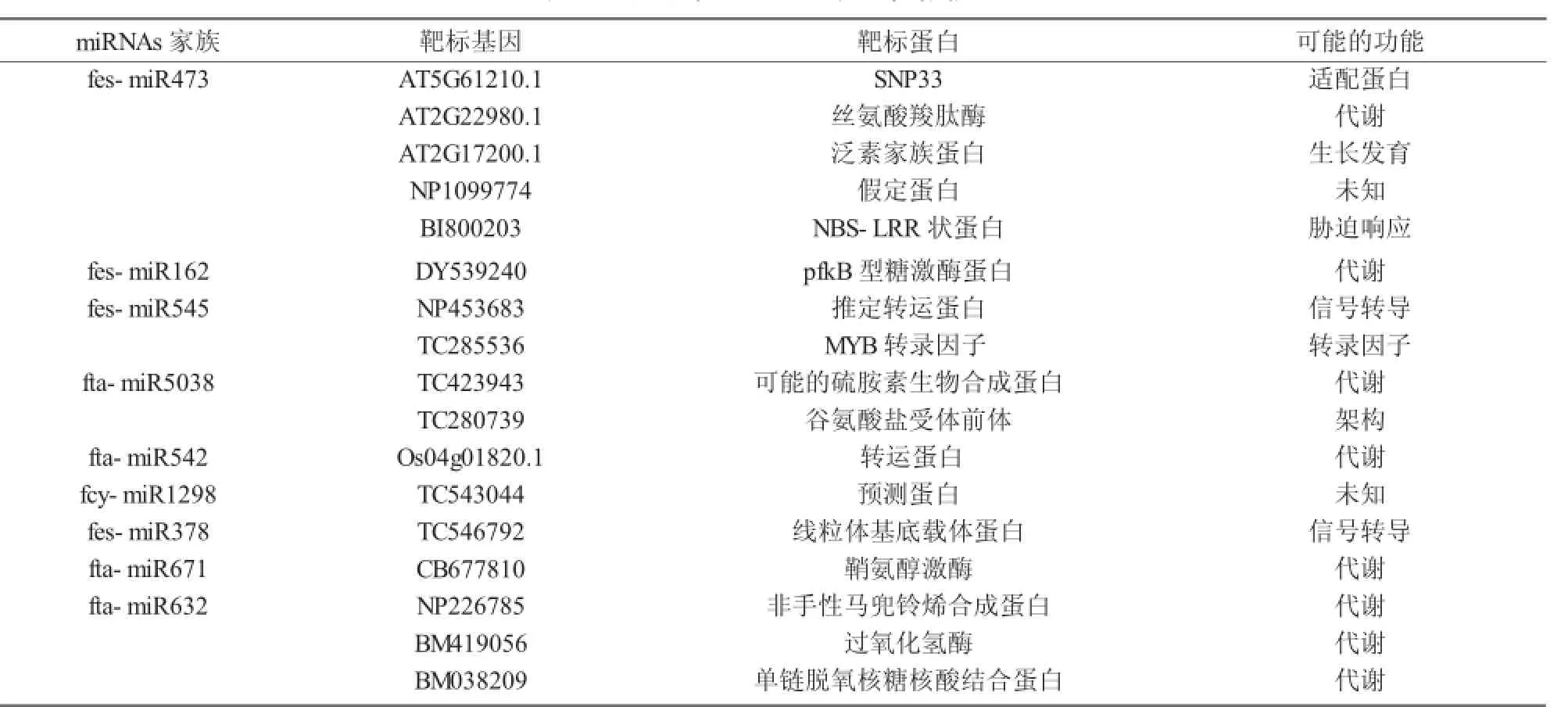
表3 预测的miRNAs家族靶标信息
2.3 靶基因及其功能预测
基于植物miRNAs与其靶位点间严格碱基互补的原则,本研究预测了荞麦miRNAs潜在的靶基因及靶位点[20],并通过蛋白质序列比对预测了靶基因的功能[27]。本研究通过ESTs从荞麦的9个miRNA家族中获得了17个靶基因。在fcy-miR2916和fta-miR3979中未预测到任何靶位点及靶基因。fes-miR473预测到的靶基因最多(5个靶基因)。预测靶基因的功能包括代谢、生长发育、胁迫响应、信号转导等方面。
9个靶基因与植物代谢过程相关,如NP226785编码非手性马兜铃烯合成酶[39],DY539240编码pfkB型糖激酶蛋白[40],AT2G22980.1编码丝氨酸羧肽酶[41]。这几个酶蛋白在植物代谢中均起着十分重要的调控作用。另外,本研究也预测了1个由AT2G17200.1编码的泛素家族蛋白,与生物体的蛋白质降解、细胞分裂、发育、免疫和其他一些复杂的生理过程紧密相关[42]。由BM038209编码的单链脱氧核糖核酸结合蛋白是DNA复制过程中的一种重要蛋白[43]。更多关于fta-miR632家族基因的研究将有助于更好地了解真核细胞DNA复制的调控机制。值得注意的是,1个由BI800203编码的与生物胁迫响应相关的靶基因也被预测到,发现该基因序列与抗病基因家族(NBS-LRR)中的亮氨酸重复保守区十分相似[44],这对于深入了解荞麦的抗病机制有重要意义。其他靶基因可能与生物体的转录和信号转导等有关(表3)。
2.4 生物信息学预测荞麦miRNAs评价
以生物信息学方法预测miRNAs及靶标较其他方法简便且成本低,适用于从那些没有任何miRNAs信息的物种中获取miRNAs。但是也存在不足。首先是已报道的相似RNA序列较少会导致较低的筛选率。本研究中被筛选出的miRNAs所占比例仅为1.1%,低于玉米的5%,原因就在于玉米已报道的相似序列较多[45]。因此,对于荞麦miRNAs的深度挖掘,该种属的高通量测序工作仍是十分必要的。其次,信息学手段预测到的miRNAs还需要进一步的试验验证。如反转录实时定量PCR,RNA干扰实验,过表达,northern杂交等,以及进一步地分析miRNAs调控下的靶基因在不同组织、不同器官、不同发育阶段的表达和目的蛋白的功能。这些验证结果将有助于详细地了解在荞麦miRNAs是如何具体调控转录后基因表达的。
2.5 荞麦miRNAs系统进化分析
对荞麦属不同miRNA家族的前体序列进行聚类,分析不同miRNA家族成员间的系统进化关系。由图3可知,所有miRNAs被分成3组。组I含有的家族成员数最多(9个家族成员),其又可分为2个亚组。从图3还可以看出,同一家族的miRNAs通常聚在一起且遗传一致性为100%,说明了同一家族miRNAs成员间的进化保守性。不同家族成员的miRNAs通常不会聚到一起,如组Ⅱ和组Ⅲ中fta和fes-miRNA家族分别聚类在2个组中,其原因可能是:一方面预测到的miRNA基因家族成员数较少,造成比对数据范围狭窄;另一方面,可能是这些m iRNAs确实参与调控了该物种不同的生理生化活动[33]。
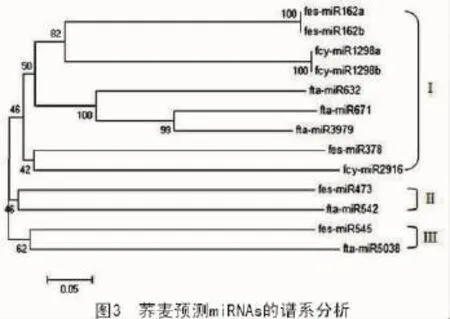
有研究报道了荞麦的起源问题[46],认为甜荞起源于我国西南部较温暖地区,其祖先是大野荞(F. megaspartanium),苦荞起源于青藏高原东部海拔较高的寒冷地区,其祖先是毛野荞(F.pilus)。图3显示,金荞麦的3个miRNAs均与甜荞的miRNAs聚为一簇,这似乎也暗示了2个物种的亲缘关系较近。考虑到金荞麦多为野生,广泛分布于与甜荞起源地气候条件相似的温暖潮湿地区[47],本研究推测金荞麦与甜荞可能有共同的祖先——大野荞,甜荞可能是大野荞经过长期进化,其中一部分种经过人为采集、种植、驯化逐渐演变成的栽培种;金荞麦则进化相对保守,未经过人为驯化或不适宜大面积种植,仅能适应原始的野生条件。
3 结论
本研究以生物信息学方法预测了荞麦属的13个miRNAs,属于11个miRNA家族。不同物种间的miRNAs序列既有保守性,也有多样性;荞麦miR1298家族与智人和家鼠同源性很高,推测其在动植物界中可能广泛存在。本研究共预测到17个靶基因,其中多数与植物的代谢调控有关。不同荞麦种间的miRNA家族系统进化分析显示,甜荞与金荞麦可能起源于共同的祖先。本研究有助于荞麦属miRNAs及靶基因表达调控机制的研究,这对荞麦的生长调控、抗病、药理、遗传和进化等方面的研究具有十分重要的意义。
[1]Chai Y,Zhang Z W.Advances in buckwheat research[M].Xi'an:North West Agriculture and Forestry University Press,2001:690-693.
[2]Jiang J F,Jia X.Sichuan Daliangshan area is one of origin region of Fagopyrumtataricum[J].Fagopyrum,1990,12(1):18-19.
[3]Li Y,Gao F,Shan F,et al.Study on the interaction between 3 flavonoid compounds and α-amylase by fluorescence spectroscopy and enzymatic kinetics[J].Journal of Food Science,2009,74(3):C199-203.
[4]Li Y,Zhou F,GaoF,et al.Comparative evaluation ofQuercetin,Isoquercetin and Rutin as inhibitors ofα-glucosidase[J].Journal ofA-gricultural and Food Chemistry,2009,57:11463-11468.
[5]Li Y,Yang P,Gao F,et al.Probing the interaction between 3 flavonoids and pancreatic lipase by methods of fluorescence spectroscopy and enzymatic kinetics[J].European Food Research and Technology,2011,233:63-69.
[6]Wang L,Yang X,Qin P,et al.Flavonoid composition,antibacterial and antioxidant properties of tartary buckwheat bran extract[J].Industrial Crops and Products,2013,49:312-317.
[7]Sensoy Í,Rosen R T,Ho C T,et al.Effect of processing on buckwheat phenolics and antioxidant activity[J].Food Chemistry,2006,99:388-393.
[8]Piao S,Li L.The actuality of produce and exploitation of Fagopyrum in China[C]//Proceeding of the 8th International Symposium on Buckwheat.Chunchon,Korea:AdvBuckwheat Res,2001:571-576.
[9]刘永文,樊燕,光德,等.荞麦芽苗研究的现状与展望[J].南方农业,2012,6(8):72-76.
[10]Lee R C,Feinbaum R L,Ambros V.The C.elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14[J].Cell,1993,75:843-854.
[11]Kim V N.MicroRNA biogenes is coordinated cropping and dicing [J].Nature Reviews Molecular Cell Biology,2005,6:376-385.
[12]Murchison E P,Hannon G J.MiRNAs on the move:miRNA biogenesis and the RNAi machinery[J].Current Opinion Cell Biology,2004,16:223-229.
[13]David P B.MicroRNAs:Target recognition and regulatoryfunctions [J].Cell,2009,136:215-233.
[14]Reinhart B J,Weinstein E G,Rhoades M W,et al.MicroRNAs in plants[J].Genes&Development,2002,16:1616-1626.
[15]Griffiths J S,Saini H K,Dongen S,et al.MiRBase:tools for microRNAgenomics[J].NucleicAcidsResearch,2007,36:D154-158.
[16]Sunkar R,Zhu J.Novel and stress-regulated microRNAs and other small RNAs fromArabidopsis[J].Plant Cell,2004,16:2001-2019.
[17]项安玲,黄思齐,杨志敏.芸苔属(Brassica)植物中MicroRNA的生物信息学预测与分析[J].中国生物化学与分子生物学报,2008,24(3):244-256.
[18]Moxon S,Jing R,Szittya G,et al.Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening[J].Genome Research,2008,18:1602-1609.
[19]Chellappan P,Jin H.Discovery of plant microRNAs and short interfering RNAs by deep parallel sequencing[J].Methods Molecular Biology,2009,495:1-12.
[20]Jones R M,Bartel D P.Computational identification of plant microRNAs and their targets,including a stress-induced miRNA[J]. Molecular Cell,2004,14:787-799.
[21]Mallory A C,Vaucheret H.Functions of microRNAs and related small RNAs in plants[J].Nature Genetics,2006,38(S):31-36.
[22]Lai E.MicroRNAs:runts ofthe genome assert themselves[J].Current Biology,2003,13:R925-936.
[23]Zhang B,Xiao P,Wang Q,et al.Identification and characterization of new plant microRNAs using EST analysis[J].Cell Research,2005,15:336-360.
[24]Xie F,Huang S,Guo K,et al.Computational identification of novel microRNAs and targets in Brassica napus[J].FEBS Letter,2007,581:1464-1474.
[25]McGinnis S,Madden T L.BAST:at the core of a powerful and diverse set of sequence analysis tools[J].Nucleic Acids Research,2004,32:W20-25.
[26]Griffiths-Jone S,Moxon S,Marshall M,et al.Rfam:annotating non-coding RNAs in complete genomes[J].Nucleic Acids Research,2005,33:D121-124.
[27]Finn R D,Coggill P,Eberhardt R Y,et al.The Pfam protein families database:towards a more sustainable future[J].Nucleic Acids Res,2016,44(D1):279-285.
[28]Du J,Wu Y,Fang X,et al.Prediction of sorghum miRNAs and their targets with computational methods[J].Chinese Science Bulletin,2010,55:1263-1270.
[29]Steffen P,Voss B,Rehmsmeier M,et al.RNAshapes:An integrated RNA analysis package based on abstract shapes[J].Bioinformatics,2006,22:500-503.
[30]张志明,宋锐,彭华,等.用生物信息学挖掘玉米中的microRNAs及其靶基因[J].作物学报,2010,36(8):1324-1335.
[31]罗晓燕,侍婷,蔡斌,等.核果类果树中microRNAs的生物信息学预测及验证[J].林业科学,2012,48(2):75-81.
[32]Aboul E H,Eiman T A,Neveen I G.Computational intelligence techniques in bioinformatics[J].Computational Biology and Chemistry,2012,47:37-47.
[33]XiangA,HuangS,YangZ.Identifyputative microRNAs in Brassica familythrough bioinformatic analysis[J].Chinese Journal ofBiochemistryand Molecular Biology,2008,24:244-256.
[34]Bartel D P.MicroRNAs:genomics,biogenesis,mechanism,and function[J].Cell,2004,116:281-297.
[35]ZhangB,Pan X,CoxSB,et al.Evidence that miRNAs are different from other RNAs[J].Cellular and Molecular Life Sciences,2006,63:246-254.
[36]宋长年,贾启东,王晨,等.32种果树microRNA的生物信息学预测与分析[J].园艺学报,2010,37(6):869-879.
[37]Lau N C,Lim L P,Weinstein E G,et al.An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans[J]. Science,2001,294:858-862.
[38]Lagos Q M,Rauhut R,Lendeckel W,et al.Identification of novel genes coding for small expressed RNAs[J].Science,2001,294:853-858.
[39]Starks C M,Back K,Chappell J,et al.Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase[J]. Science,1997,277:1815-1820.
[40]Soderlund C,Descour A,Kudrna D,et al.Sequencing,mapping,and analysis of 27,455 maize full-length cDNAs[J].PLOS Genetics,2009,5:e1000740.
[41]Felix S,Wolfgang B,Jürgen S,et al.Activities of Arabidopsis sinapoylglucose:malate sinapoyltransferase shed light on functional diversification of serine carboxypeptidase-like acyltransferases [J].Phytochemistry,2008,69:1826-1831.
[42]Nikolaos G S,Mayank M P,Angel E G,et al.Conformational dynamics and structural plasticity play critical roles in the ubiquitin recognition of a UIM domain[J].Journal of Molecular Biology, 2010,396:1128-1144.
[43]Ehn M,Ahmadian A,Nilsson P,et al.Escherichia coli single stranded DNA binding protein,a molecular tool for improved sequence quality in pyrosequencing[J].Electrophoresis,2002,23:3289-3299.
[44]Belkhadir Y,Subramaniam R,Dang J.Plant disease resistance protein signaling:NBS-LRR proteins and their partners[J].Current Opinion in Plant Biology,2004,7:391-399.
[45]Li Y,Li W,Jin Y.Computational identification of novel family members of microRNA genes in Arabidopsis thaliana and Oryza sativa[J].Acta Biochimica et Biophysica Sinica,2005,37(2):75-87.
[46]Chen Q,Sai L,Friedrich J.A study of cytology,isozyme and inter specific hybridization on the big achene group of buckwheat species(Fagopyrum,Polygonaceae)[J].Crop Sciences,2004,44:1511-1518.
[47]Kevin C,Nikolaus R.Natural selection on human microRNA binding sites inferred from SNP data[J].Nature Genetics,2006,38:1452-1456.
Prediction of microRNAs and Their Target Genes Using Bioinformatics Methods in GenusFagopyrum
GAOFan1,SONGWei2,XIE Shulian1
(1.College ofLife Sciences,Shanxi University,Taiyuan 030006,China;2.Shanxi Sports Vocational School,Taiyuan 030006,China)
MicroRNAs(miRNAs)are small molecular non-coding RNAs which play important roles in post-transcriptional gene regulation.Discovering and identifying newmiRNAs is tedious using traditional experimental methods.Searching for miRNAs from RNA databases with bioinformatics methods can improve the efficiency of seeking new miRNAs and their targeted genes greatly.A large number ofmiRNAs are reported in plants,but research on the genusFagopyrumhas returned no results so far.After prediction searching expressed sequence tags(ESTs)and genome survey sequences(GSSs)from buckwheat with miRNAs reported from all plants,we predicted 13 miRNAs in 11 miRNA families using strict screening.The predicted miRNAs sequences embodied conservative in different plants,but the number of the miRNAs distribution embodied diverse.Seventeen potential targets were predicted by our analysis of the predicted miRNAs with mRNAs and ESTs in plants.The targeted proteins were necessary in metabolism,growth and development,stress response,signal transduction and so on,which suggested the miRNAs played an essential role in life processes.Phylogenetic analysis suggested that miRNAs evolution ofF.cymosummight be related withF.esculentum's.Discovered and identified newmiRNAs and their targeted genes in genusFagopyrumfromthe known databases with bioinformatics methods should be feasible.
MicroRNA(miRNA);bioinformatics;Fagopyrum;target gene
Q752
A
1002-2481(2016)09-1237-07
10.3969/j.issn.1002-2481.2016.09.01
2016-04-05
山西省煤基重点科技攻关项目(FT201402-15)
高帆(1985-),男,山西长治人,实验师,博士,主要从事生物信息研究工作。谢树莲为通信作者。
猜你喜欢
今日农业(2022年13期)2022-09-15
天津医科大学学报(2021年1期)2021-12-05
今日农业(2021年3期)2021-12-05
现代畜牧科技(2021年4期)2021-07-21
天津医科大学学报(2019年3期)2019-08-13
小小说月刊(2018年12期)2018-12-13
中国博物馆(2018年2期)2018-12-05
黄河·黄土·黄种人(华夏文明)(2017年3期)2017-08-22
现代检验医学杂志(2016年5期)2016-08-20
现代检验医学杂志(2015年4期)2015-02-06
