
柱前衍生化-气相色谱法测定水产品中的游离甲醛
2017-01-04 03:02汤施展黄丽陈中祥白淑艳张芳梅吴松覃东立牟振波
中国渔业质量与标准 2016年2期
汤施展,黄丽,陈中祥,白淑艳,张芳梅,吴松,覃东立,牟振波
(中国水产科学研究院黑龙江水产研究所,黑龙江 哈尔滨 150070)
柱前衍生化-气相色谱法测定水产品中的游离甲醛
汤施展,黄丽,陈中祥,白淑艳,张芳梅,吴松,覃东立,牟振波*
(中国水产科学研究院黑龙江水产研究所,黑龙江 哈尔滨 150070)
应用柱前衍生化-气相色谱(GC)法检测水产品中游离甲醛含量。野生鲤、鳙、鲢、草鱼、鲫和乌苏里白鲑等样品经超声萃取、衍生化、正己烷提取,用HP-5毛细管色谱柱分离,电子捕获检测器进行检测,外标法定量。结果表明,使用优化改进后的方法测定的游离甲醛含量在0.1~50 μg/L范围内,标准曲线线性拟合度r2= 0.999,线性良好;在2.0、5.0、10.0 μg/kg 3个水平下加标回收率为83.80%~103.15%,相对标准偏差小于5.0%。方法检出限为0.38 μg/kg,定量限为0.50 μg/kg。本研究调查了查干湖和黑龙江中的野生鱼类中游离甲醛的本底含量为1.67 ~7.66 mg/kg,地区与种间差异不显著(P>0.05)。优化后的方法操作简便,分析灵敏度高,稳定性好,可满足水产品中微量游离甲醛的检测需要,对开展水产品中甲醛本底含量及安全限量的研究具有重要的现实意义。[中国渔业质量与标准, 2016, 6(2):51-56]
水产品;游离甲醛;气相色谱法;柱前衍生化;2,4-二硝基苯肼
甲醛别名蚁醛,35%~40%的甲醛水溶液俗称福尔马林。研究表明甲醛具有刺激作用、致敏作用、免疫毒性、神经毒性、生殖毒性、遗传毒性和致癌性等危害[1]。中国有毒化学品优先控制名单上甲醛位居第二,国际癌症研究机构(IARC)将其确定为人类致癌物(Group1)和重要的环境污染物[2]。然而,由于甲醛是活泼的烷化剂,可以使蛋白质变性,对寄生虫、藻类、真菌、细菌、芽孢和病毒均有较强的杀菌效果,因此在水产养殖过程中被广泛用于鱼类和甲壳类的疾病防治。而且,甲醛具有漂白、防腐作用,能够延长食品保质期,增加食品持水性和韧性,许多不法商贩常在水产品或水发产品中违规添加甲醛,水产品中甲醛污染问题已引起社会广泛关注[3]。此外,甲醛作为一种内源性的代谢中间产物在水产品中普遍存在,其本底含量因水产品的生物学类别而异,另有研究表明有些水产品经冷冻保鲜后,由于氧化三甲胺酶的作用,甲醛含量会明显升高[3-4]。因此针对种类繁多且基质成分复杂的水产品,建立甲醛快速、灵敏的定性定量检测方法显得尤为重要。
水产品中甲醛主要以游离态、可逆结合态和不可逆结合态的形式存在,水浸法和蒸馏法提取可得到游离态和各种结合态的甲醛。结合态甲醛主要影响水产品的感官性状,而游离态甲醛是危害消费者身体健康的主要影响因子,因此有研究者建议测定食品中游离态的甲醛含量替代食品中总甲醛含量[4-5]。目前,国内外报道的检测甲醛的方法主要有分光光度法[6-9]、色谱法[10-13]、酶法[14]、荧光法[15]、电化学传感器法[16]和极谱法[17],其中只有荧光法涉及游离态甲醛含量检测,其他均为总甲醛含量检测,适用于水产品中甲醛检测的方法主要有紫外法、液相法和气相法。中国水产行业标准(SC/T 3025—2006)中规定的紫外法和液相法的检出限分别为0.5和0.2 mg/kg。上述方法大都存在样品前处理过程复杂、检出限高、抗干扰能力差等不足。为解决上述问题,本研究采用2,4-二硝基苯肼柱前衍生化-气相色谱法检测水产品中游离甲醛,并对游离甲醛的萃取和检测条件进行了优化与探讨,建立了准确、高效、抗干扰性强的检测水产品中游离甲醛的气相色谱法,以期为水产品中甲醛安全限量的制定、游离甲醛含量的检测及其代谢规律的研究提供技术支持。
1 材料与方法
1.1 材料与仪器
1.2.1 试剂
甲醛标准储备液,质量浓度为1 000 mg/L,由 AccuStandard公司生产;2, 4-二硝基苯肼(DNPH),由上海安普科学仪器有限公司提供,纯度(HPLC)≥98.0%。DNPH衍生化试剂配制:分别称取0.010 0、0.050 0、0.100 0、0.200 0、0.250 0、0.300 0和0.500 0 g的2, 4-二硝基苯肼溶解于25 mL浓盐酸中,用超纯水定容到100 mL,配置成质量浓度为100、500、1 000、2 000、2 500、3 000和5 000 mg/L的2, 4-二硝基苯肼溶液。正己烷(Merck公司)为农残级;实验用水为超纯水电阻率为18.0 Ω。
1.1.2 仪器
7890 B气相色谱仪(美国Agilent公司)配备电子捕获检测器(ECD检测器);电子天平(梅特勒-拖利多公司);电子恒温水浴锅(天津市泰斯特仪器有限公司);KQ-700E型超声波清洗器(昆山市超声仪器有限公司);超纯水系统(Milli-Q公司);高速离心机(美国热电公司)。
1.2 实验方法
1.2.2原理
甲醛在酸性条件下与2,4-二硝基苯肼反应生成2,4-二硝基苯腙,经正己烷萃取后用电子捕获检测器-气相色谱仪检测,间接测得水产品中游离甲醛含量。
1.2.3 仪器条件
色谱柱Aglient: HP-5 (5% Phenyl Methyl Siloxane),30 m × 320 μm ×0.25 μm;进样口温度为200 ℃;载气为高纯氮气;流速为1.0 mL/min;尾吹流量为 60 mL/min;柱温箱初始温度为100 ℃,阶升20 ℃/min至240 ℃;检测器温度为300 ℃;不分流进样;进样量为 1 μL。
1.2.4 标准曲线绘制
准确吸取一定量的甲醛标准储备液,用纯水逐级稀释,得到质量浓度梯度为0.10、0.25、0.50、1.00、2.50、5.00、10.00、25.00、50.00 μg/L的甲醛工作曲线,然后加0.5 mL 1 000 mg/L 2,4-二硝基苯肼溶液,在60 ℃恒温水浴锅中反应30 min,取出立即用流水冷却,加10 mL正己烷萃取,取1.0 mL萃取液待测。同时用纯水做空白。
1.2.5样品测定
水产品取背部肌肉组织约200 g左右,置于高速匀浆机中匀浆,制成均匀的糜状样品,装入洁净的样品袋内,-20 ℃冰箱保存。测定前,样品在4 ℃冰箱中解冻,准确称取1 g左右样品,加10 mL纯水,涡旋3 min,超声20 min,5 000 r/min离心3 min,取5 mL上清液,依照1.2.3方法衍生化后,待测。
2 结果与讨论
2.1 超声波萃取时间的确定
甲醛沸点低,易溶于水。取同一质量浓度0.1 mg/L甲醛标准溶液,在相同实验条件下分别超声10、20、30、45、60和90 min,重复3次。检测衍生物生成量的结果见图1,萃取20 min时衍生物峰面积最大,说明超声萃取20 min游离甲醛的提取效果最佳。

图1 超声时间对衍生物生成量的影响Fig.1 The effects of ultrasonic time on the derivative production(n=3)
2.2 衍生化时间的选择
在甲醛与2,4-二硝基苯肼溶液衍生化生成2,4-二硝基苯腙的反应中,如果衍生化时间过短,衍生反应不完全;而衍生化时间过长,会因甲醛挥发造成较大误差。为了解衍生化时间对衍生物生成量的影响,本研究取同一浓度的一组甲醛溶液各5 mL,分别加入0.5 mL 1 000 mg/L 2,4-二硝基苯肼溶液,于60 ℃下水浴衍生化0、10、20、30、40、50、60、90和120 min。取出后在流水中快速冷却,然后用10 mL正己烷分3次(3、3和4 mL)提取,涡旋3 min,5 000 r/min离心3 min,合并提取液,分别定容至10 mL,并取1 mL上机检测,实验重复3次。样品残留液再用10 mL正己烷重复提取1次,用于样品提取效率分析。结果如图2所示,衍生化40 min时,衍生物生成量最大,随后逐渐下降,表明40 min时衍生化反应完全,随着衍生化时间延长衍生物生成量有损失。确定衍生化时间为40 min。从提取情况来看,经过3次提取后,样液中游离甲醛衍生物提取率达到96.67%~97.91%。

图2衍生时间对衍生物生成量的影响Fig.2 The effects of derivatization time on the derivative production(n=3)
2.3 衍生化试剂浓度的选择
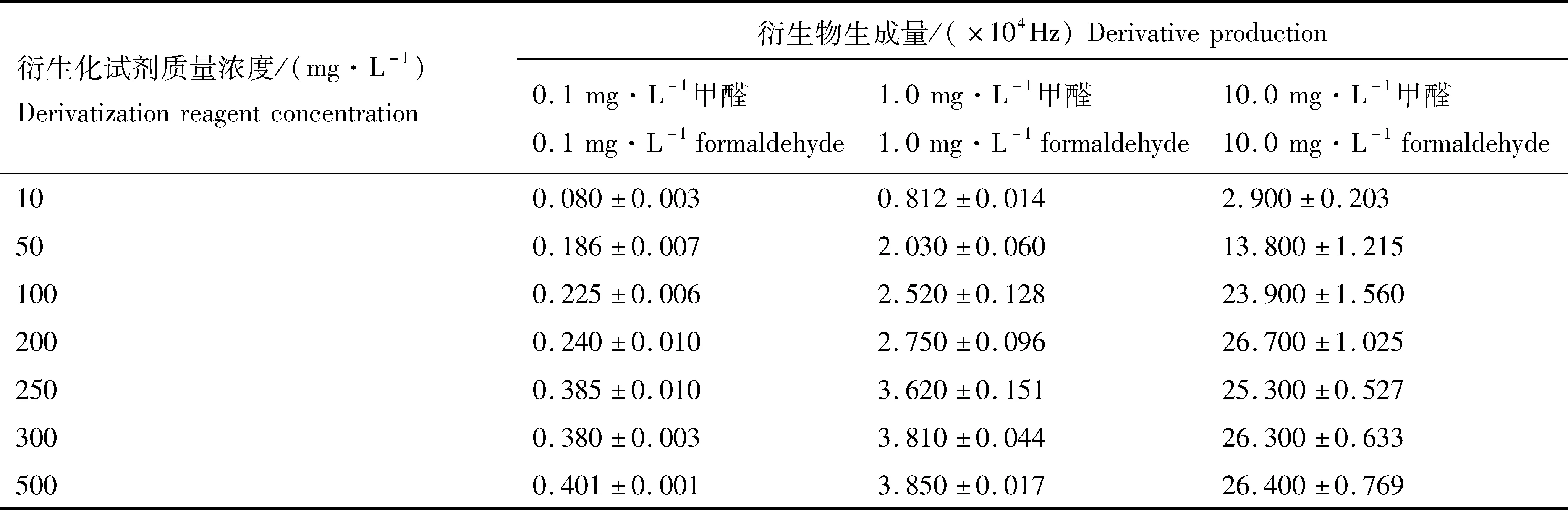
衍生化试剂浓度对衍生物生成量的影响很大。由于衍生化试剂本身也出色谱峰,如果用量过大,不仅造成浪费,而且会引起色谱柱过载,影响目标峰分离与定量。衍生化试剂量过小,则会导致衍生化不完全。设计0.1、1.0和10 mg/L 3组浓度梯度的甲醛加标样,分别加入0.5 mL衍生化试剂,样液中衍生化试剂质量浓度分别为10、50、100、200、250、300和500 mg/L,实验重复3次。衍生化后衍生物生成量结果见表1。
从表1可知,0.1和1.0 mg/L甲醛溶液在衍生化试剂质量浓度为250 mg/L时,衍生物生成量趋于稳定;10 mg/L甲醛溶液在衍生化试剂质量浓度为200 mg/L时,衍生物生成量最大。可以看出,3个质量浓度的甲醛溶液在衍生化试剂浓度低于250 mg/L时,衍生物峰面积随着衍生化试剂质量浓度的增加而递增,衍生化试剂质量浓度为250~500 mg/L时反应趋于稳定。为了衍生化完全,确定最佳衍生化试剂质量浓度为300 mg/L。
表1 衍生化试剂质量浓度对衍生物生成量的影响
Tab.1 The effects of derivatization reagent concentration on the derivative production
n=3
2.4 分离条件的确定
前期实验中,根据甲醛衍生物的特性,分别采用中等极性色谱柱DB-1701和非极性色谱柱HP-5进行实验,以HP-5的分离效果最佳;在色谱分离条件优化时,柱温和载气流速对目标峰分离及检测稳定性影响很大。通过改变载气流速实验研究发现,载气流速为1.0、1.5、2.0、2.5、3.0、3.9 mL/min时,相应的出峰时间分别为8.78、7.74、7.21、6.87、6.62、6.30 min。为了提高检测效率,在载气流速为3.9 mL/min的基础上,将升温程序改为:初始温度为120 ℃,保持0 min;以25 ℃/min升温速率将温度爬升至240 ℃,保持3 min;检测器温度为300 ℃,进样口温度为200 ℃。目标峰出峰时间缩短为4.55 min,总检测时间为7.8 min,极大地提高了检测效率。
2.4.1 标准曲线
按1.2.3配制的系列标准曲线,以峰面积为纵坐标,浓度为横坐标,测得回归方程为Y=mX+b,其中,m=619.599 73,b=-220.829 12,线性相关系数r为0.999 69。线性范围为0.1~50 μg/L。重复测定0.3 μg/L样品7次,标准偏差S=0.006,参照美国环境保护局(EPA)推荐方法[18],以取样量1.00 g计,本方法对样品的检出限为0.38 μg/kg(以S/N>3计),定量限为0.50 μg/kg(以S/N>10计)。为满足实验需要,绘制3条标准曲线(表2)和10 μg/L甲醛标准溶液衍生物色谱图(图3)。
表2 不同浓度范围甲醛标准曲线方程
Tab.2 Calibration curves of formaldehyde in different concentration range


图3 甲醛标准溶液衍生物色谱图Fig.3 Chromatogram of standard formaldehyde derivatization
2.4.2 方法准确度和精密度
加标回收实验中,选取查干湖鳙作为空白加标样品,分别加入质量浓度为2.0、5.0、10.0 μg/mL的甲醛标准溶液各1.0 mL,按照1.2.3方法步骤1 d内测定3次,每样平行测定6次,计算日内精密度,1周连续测定3次,每样平行测定6次,计算日间精密度,得到数据见表3。总体加标回收率为83.80%~103.15%,日内和日间精密度的相对标准偏差(RSD)均小于5.0%,表明该方法测定的精密度高,稳定性好,适用于水产品中微量游离甲醛的快速测定。
表3 鳙甲醛加标回收率和相对标准偏差
Tab.3 The recoveries and relative standard deviation of spiked formaldehyde inAristichthysnobilis
n=6

表4 野生鱼类甲醛本底含量
n=3

2.5 样品检测
本研究对查干湖和黑龙江野生鱼类肌肉中游离甲醛含量进行测定,样品前处理采用优化后的方法,以1.0 mL/min流量模式进行上机检测,为保证检测结果准确性,每个样品平行测定3次,同时进行加标回收实验,样品加标回收率为90.50%,样品检测结果见表4。可以看出,6种被检测的淡水鱼甲醛含量在1.67 mg/kg(查干湖草鱼)至7.66 mg/kg(黑龙江乌苏里白鲑)之间,地区与种间差异不显著(P>0.05)。从检测结果来看,常见野生淡水鱼类自身含有一定量的甲醛。刘淑玲[19]采用乙酰丙酮分光光度法检测中国淡水鱼类的甲醛含量范围为0.25~2.96 mg/kg,中位值为0.25 mg/kg,低于本研究结果。分析其原因,一方面可能是由于活鱼甲醛含量低于冰鲜样品,冷冻样品随时间延长甲醛含量上升[4];另一方面也可能由于其采用乙酰丙酮分光光度法检测的检出限为0.50 mg/kg,低于检出限的样品均按0.25 mg/kg计算[19],导致所检测样品含量整体偏低。
另外,同一批次样品的甲醛衍生液在-20℃冷冻保存15 d后重新测定样品,发现其含量相差不大,表明该衍生物在此条件下保存比较稳定。本研究同时采用常规蒸馏法测定了查干湖鳙鱼肌肉中总甲醛含量,对比发现,游离甲醛占总甲醛的40%左右,这与Tai等[5]的研究结果相近。
3 结论
与SC/T 3025—2006《水产品中甲醛的检测》[6]方法相比,本研究在前处理上采用超声提取法缩短了提取时间,克服了蒸馏操作费时费力的缺点,通过HP-5毛细管色谱柱分离电子捕获检测器检测,提高了检测灵敏度,柱前衍生化法降低了样品基质干扰。本方法具有检出限低、稳定性强、回收率高、出峰时间稳定、信号响应值高和操作简便等优点,为水产品中甲醛残留及内、外源甲醛来源的研究提供技术支持。另外,本研究检测了黑龙江和查干湖野生水产品中游离甲醛的本底含量为1.67~7.66 mg/kg,为水产品中甲醛标准限量的制定提供基础数据。而关于水产品中体内和体外甲醛的研究工作,有待于下一步开展。
[1] 权可艳, 李正军. 常用鱼药使用手册 [M]. 成都:四川科学技术出版社, 2011: 61-63.
[2] IARS/WHO. Classifies Formaldehyde As Carcinogenic to Humans [R]. Geneva:World Health Organization, 2004.
[3] 覃梅, 杨健. 水产品中甲醛含量的检测研究 [J]. 化学工程与装备, 2013(4): 166-169.
[4] 谢雯雯, 熊善柏.水产品中甲醛的残留及控制[J].农产品加工·创新版,2013(1):20-23.
[5] Tai S Y, Tzu C L, Ching C C, et al. Analysis of free and bound formaldehyde in squid and squid products by gas chromatography-mass spectrometry [J]. J Food Drug, 2013(21): 190-197.
[6] 周德庆,马敬军,李晓川. SC/T 3025—2006 水产品中甲醛的检测 [S]. 北京:中国标准出版社, 2006: 1-6.
[7] Bunkoed O, Davis F, Kanatharanaa P, et al. Sol-gel based sensor for selective formaldehyde determination [J]. Anal Chim Acta, 2010, 659: 251-257.
[8] Teshima N, Fernández S K M, Ueda M, et al.Flow injection spectrophotometric determination of formaldehyde based on its condensation with hydroxylamine and subsequent redox reaction with iron(III)-ferrozine complex [J]. Talanta, 2011, 84:1205-1208.
[9] Afkhami A, Bagheri H. Preconcentration of trace amounts of formaldehyde from water, biological and food samples using an efficient nanosized solid phase, and its determination by a novel kinetic method [J]. Microchim Acta, 2012, 176(1/2):217-227.
[10] Wang H, Ding J, Du X, et al.Determination of formaldehyde in fruit juice based on magnetic strong cation-exchange resin modified with 2,4-dinitrophenylhydrazine [J]. Food Chem, 2012, 131(1):380-385.
[11] Xu X, Su R, Zhao X, et al. Determination of formaldehyde in beverages using microwave-assisted derivatization and ionic liquid-based dispersive liquid-liquid microextraction followed by high-performance liquid chromatography [J]. Talanta, 2011, 85(5):2632-2638.
[12] Nageswari A, Krishna Reddy K V S R , Mukkanti K. Low-level quantitation of formaldehyde in drug substance by HPLC-UV[J]. Chromatographia, 2012, 75(5/6):275-280.
[13] Shin H, Lim H. Simple determination of formaldehyde in fermented foods by HS-SPME-GC/MS[J]. Int J Food Sci Tech, 2011, 47(2):350-356.
[14] Vladimir S, Olha D, Halyna K. Alcohol oxidase- and formaldehyde dehydrogenase-based enzymatic methods for formaldehyde assay in fish food products[J].Food Chem, 2011, 127(2):774-779.
[15] Sáenz M, Alvarado J, Pena-Pereira F, et al. Liquid-phase microextraction with in-drop derivatization combined with microvolume fluorospectrometry for free and hydrolyzed formaldehyde determination in textile samples[J]. Anal Chim Acta, 2011, 687:50-55.
[16] Marzuki N I, Bakar F A, Salleh A B, et al. Development of electrochemical biosensor for formaldehyde determination based on immobilized enzyme[J]. Int J Electrochem Sci, 2012, 7(7):6070-6083.
[17] 戴红, 吴颉, 朱正鑫, 等. 伏安极谱法测定兔毛皮中可萃取甲醛含量的研究[J]. 皮革科学与工程, 2014 (6):63-67.
[18] Childress C J O, Foreman W T, Connor B F, et al. Newreporting procedures based on long-term method detection levels and some considerations for interpretations of water-quality data provided by the U.S. geological survey national water quality laboratory [M]. Reston,Virginia:Bibliogov,1999:19.
[19] 刘淑玲. 水产品中甲醛的风险评估与限量标准研究[D]. 青岛:中国海洋大学, 2009.
Quantification of free-formaldehyde in aquatic products by gaschromatography coupled with pro-column derivatization
TANG Shizhan,HUANG Li, CHEN Zhongxiang, BAI Shuyan, ZHANG Fangmei, WU Song, MOU Zhenbo*
(Heilongjiang River Fishery Research Institute Chinese Academy of Fishery Sciences, Harbin 150070, China)
In this study, the gas chromatography (GC) coupled with pro-column derivatization method was employed to determine free-formaldehyde in aquatic products. Samples ofCyprinuscarpio,Aristichthysnobilis,Hypophthalmichthysmolitrix,Ctenopharyngodonidellus,CarassiusauratusandCoregonusussuriensiswere subjected to ultrasonic extraction, derivatization, n-hexane extraction, and then purified by HP-5 chromatographic column and determined by external standard method with electron capture detector (ECD). The results showed that the optimized method had a good linearity (r2=0.999) when the measured free-formaldehyde was 0.1-50 μg/L, and the limit of detection was 0.38 μg/kg with the relative standard deviations no more than 5.0%. The recoveries of formaldehyde among three different levels (2.0, 5.0, 10.0 μg/kg, respectively) were in the range of 83.80%~103.15%. The baseline of free-formaldehyde in wild fish species from the Chagan Lake and Heilongjiang River were investigated, which ranged from 1.67 - 7.66 mg/kg and without significant differences in regions and species (P> 0.05). The optimized analytical method was rapid and accurate, and suitable for the determination of free-formaldehyde in aquatic products.[Chinese Fishery Quality and Standards, 2016, 6(2):51-56]
aquatic products; free-formaldehyde; gas chromatography; pro-column derivatization; 2, 4 - dinitrophenyl hydrazine
MOU Zhenbo, mouzb1962@163.com
2015-09-08;接收日期:2015-12-21
水产品中未知危害因子识别与已知危害因子跟踪评估(GJFP201501001)
汤施展(1987-),男,学士,研习员,研究方向为水产品质量与安全,tsz2007@163.com
牟振波,研究员,研究方向为水产养殖,mouzb1962@163.com
S94
:A
:2095-1833(2016)02-0051-06
猜你喜欢
今日农业(2021年4期)2021-11-27
今日农业(2021年15期)2021-11-26
青岛科技大学学报(自然科学版)(2021年3期)2021-06-09
分析仪器(2020年4期)2020-09-03
渔业致富指南(2019年21期)2019-11-21
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
环境监控与预警(2016年5期)2016-04-18
合成化学(2015年10期)2016-01-17
