
利用超级集群分离分析法鉴别大豆增变基因
2016-12-15 08:09:45黄志刚陈吉宝
西北农林科技大学学报(自然科学版) 2016年12期
常 玮,王 娟,黄志刚,陈吉宝
(南阳师范学院 农业工程学院,河南 南阳 473061)
利用超级集群分离分析法鉴别大豆增变基因
常 玮,王 娟,黄志刚,陈吉宝
(南阳师范学院 农业工程学院,河南 南阳 473061)
【目的】 利用大豆基因组Wm82.a2.v1及HapMap数据来鉴别大豆中可能存在的增变基因。【方法】 将大豆HapMap数据(包含19 652份大豆种质在52 041个位点上的分型结果)预处理后,利用改进的超级集群分离分析法,选取滑动窗口大小、步长及阈值大小为参数,对预处理后的大豆种质数据进行分析,测算大豆点突变比率及突变热点区数目,并将点突变比率及突变热点区数目与分子标记进行关联性分析,从强关联区内挖掘候选增变基因,用大豆基因组Wm82.a2.v1对其功能进行初步推测。【结果】 大豆HapMap数据预处理后,15 391份大豆种质点突变比率为0.089~0.531,平均变异率为0.261;突变热点区数目为5~1 324个,平均每份种质包含347.8个突变热点区。超级集群分离分析结果表明,Gm16上的29 153 474-30 604 603 bp和Gm17上的12 133 293-12 147 725 bp片段为与点突变比率和突变热点区数目2个表型同时存在强关联的区域;其中Gm16上的Glyma16g26440.1和Gm17上的Glyma17g15420.1同属于nudix水解酶基因家族,推测其与基因组突变有关。【结论】Glyma16g26440.1和Glyma17g15420.1 2个nudix水解酶基因家族成员都与点突变比率和突变热点区数目存在强关联,可作为大豆基因组候选增变基因。
大豆;突变热点;增变基因;超级集群分离分析;nudix水解酶基因
大豆是重要的油料作物和植物蛋白的主要来源。目前大豆育种的常规方法是将两亲本进行杂交,再通过后代连续的自交选择获得新的大豆种质。由于大豆异花授粉难度很大,与玉米等作物相比,无法产生较为丰富的变异类型,而通过化学或物理的方式进行人工诱变,可以获得较为丰富的变异类型。但限于突变率较低的原因,人工诱变仍未能获得广泛应用。因此通过研究增变基因来提高大豆突变率,将会大大促进大豆突变育种的发展。
在生物学中,突变是指细胞中的遗传物质(如染色体DNA、细胞质DNA以及其他遗传单元)发生改变的自然过程。它包括单个碱基改变所引起的点突变或多个碱基的缺失、重复和插入。突变通常是由辐射、化学诱变剂或一些可移动遗传原件,如转座子的插入及删除引起的DNA序列改变[1-2]。从理论上讲,DNA分子上每一个碱基都可能发生突变,但实际上突变部位并非完全随机分布,因为DNA分子上的各个部分有着不同的突变频率,某些部位的突变频率明显高于平均数,这些部位就称为突变热点[3]。对突变热点的研究,不仅可以揭示突变产生的机制,还可以阐明某一目标蛋白功能域(Functional domains)所具有的功能[4-5]。p53蛋白是迄今为止研究最多的一种抑癌蛋白,在调控机体代谢、生殖方面以及肿瘤发生中发挥着重要作用。TP53突变在肿瘤发生中比较常见,且不同位点的点突变产生了多种形式的突变p53蛋白[6-7]。不仅如此,植物基因组上也存在在着突变热点。例如,在大豆中,脂氧合酶基因Lox1和Lox2存在突变热点区[8]。此外,对人类及大豆等HapMap数据的分析发现,突变位点的非随机分布还表现在同一物种的不同个体间,例如大豆种质PI479750所包含的突变位点数目是PI416792的6倍[9]。
目前,对于突变热点形成的原因仍未完全明了。有研究指出,在人类基因的突变热点中,C∶G二核苷酸更容易发生突变,这是因为5-甲基胞嘧啶(MeC)经脱氨基作用就转变成胸腺嘧啶(T),这样,在DNA修复系统不完善的情况下,G∶C配对就可能变成G∶T错配,从而引起基因突变[10-11]。还有证据表明,在短的连续重复序列处容易发生插入或缺失突变,如鼠伤寒沙门氏菌hisD3052的基因序列上CGCGCGCG重复区域易发生二碱基对缺失[12];其产生机制可能是由于短的同源重复序列之间发生了重组[13],或DNA聚合酶在短重复序列之间发生了滑移[14]。此外,突变热点还与突变剂种类有关,如Lox2基因第8外显子上存在γ射线敏感位点,而在人工培育的哺乳动物细胞中,TP53上含有MeC的嘧啶二聚体更容易在紫外线照射下发生突变,从而诱发皮肤癌[8,15]。在上述研究的基础上,Rogozin等[16]对突变热点周围序列特征进行了详细论述,总结了18种易突变模体(Mutable motif)。
除DNA结构的影响外,在生物体内,还存在着一种与突变发生和抑制相关的基因,称为增变基因(Mutators)。增变基因通常与DNA修复有关,例如人类的DNA聚合酶β和载脂蛋白B mRNA编辑酶(Apolipoprotein B mRNA editing enzyme,catalytic polypeptide-like,APOBEC)[17-18]。Jonathan等[19]总结了原核生物和真核生物中的增变基因,并阐述了其在逆境胁迫中的功能。在植物中也有类似功能的增变基因,如拟南芥中的AtMSH2已经被证实具有稳定基因组结构和增变的效应[20]。但大豆中还未发现此类基因。
采用集群分离分析法(Super Bulked Segregant Analysis,BSA)进行功能基因挖掘已经积累了许多成功的案例[21]。然而,由于传统的BSA方法只能对由少数个体组成的集群进行分析,因此检测效力较低,且容易产生假阳性结果。例如,在传统BSA研究中,由10个个体组成的混合群体,如果其中某个个体的基因型与其他个体基因型不同,则不能判定该位点是否与性状相关联。而超级集群分离分析法(Super Bulked Segregant Analysis,Super-BSA)是基于HapMap数据进行的,混合群体内每个个体的基因型均为已知,因此可以根据某位点不同基因型的比率来进行关联性分析,提高了检测效力[22]。
本研究以大豆基因组Wm82.a2.v1及HapMap数据为基础,采用Super-BSA的理论及算法,对大豆不同个体间单核苷酸多态性(Single nucleotide polymorphism,SNP)热点区及数目差异产生的机制进行遗传分析,同时挖掘具有增变基因效应的候选基因,以期为大豆突变育种研究提供参考。
1 材料与方法
1.1 数据来源及预处理
大豆基因组Wm82.a2.v1,用于基因组定位及功能基因注释,下载于phytozome网站(http://phytozome.jgi.doe.gov/,2014-01-24更新)[23]。大豆HapMap数据(包含19 652份大豆种质在52 041个位点上的分型结果)来源于SoyBase(http://soybase.org/snps)[9],用于数据挖掘。应用Plink version 1.07的质量控制功能来进行大豆HapMap数据的预处理[24],处理标准如下:剔除频率(Minor Allele Arequency,MAF)小于1%的稀有位点,缺失超过5%、且违背Hardy-Weinberg平衡(P<1×10-6)的SNP位点,以及缺失基因型、杂合基因型和未知基因型总数大于10%的种质。
1.2 数据标准化及点突变比率的计算
采用Perl脚本对经质量控制后的大豆HapMap数据进行标准化处理,计算每份大豆种质的点突变比率(R_snp)。数据标准化方法如下:剔除杂合基因型及未知基因型,统计每个位点上不同基因型数目,并将其中数目最多的基因型作为背景,记作‘0’;另外一种基因型作为突变,记作‘1’。
在此基础上计算每份大豆种质的点突变比率,计算方法如下:
R_snp=Sum1/(Sum1+Sum0)。
式中:Sum1和Sum0分别为某份大豆种质在所有剩余位点中所包含的基因型为‘1’和‘0’的数目。
1.3 突变热点区的确定
点突变热点区的确定采用滑动窗口法进行。首先以每份大豆种质为单位,根据标准化的数据及不同位点的位置信息,利用R程序统计相邻两位点间距大小的分布情况,并绘制概率密度函数曲线,同时利用quantile()函数求得累积概率为0.95时所对应的间距大小,并以此为窗口大小。以5%窗口大小为步长,并以该窗口内所包含SNP位点数期望值的倍数作为热点区判断的阈值,计算方法如下:
Thotspot=N+Wlength/μlen。
其中:Thotspot为热点区判断的阈值,Wlength为窗口大小,μlen为所有位点间距的均值,N取自然数。
根据N值及窗口大小和步长,确定不同阈值条件下每份大豆种质所包含的热点区的数目。将不同阈值条件下所得到的突变热点区数目与点突变比率进行简单相关分析,满足相关系数>0.5的最大自然数即为最终的N值,并依此求得Thotspot。
1.4 点突变比率及突变热点区数目的超级集群分离分析
以点突变比率及热点区数目为表型,设这2个表型在大豆种质群体中的分布符合高斯混合模型(Gaussian Mixture Model,GMM),采用R程序中的EM算法来进行GMM的参数估计,再根据参数估计的结果计算混合分布两侧各组分所占比例为90%的分位数,并以此为依据进行分群。
在上述分群的基础上,参考Super-BSA的理论方法,采用改进的算法进行点突变比率及热点区数目与分子标记的关联性分析[22]。具体做法为:分别计算2集群(L集群和H集群)内每个位点上基因型‘1’和‘0’的比值R,分母为0时,比值计为1 000;根据分群标准,统计L集群内R大于9、同时H集群内R小于1/9,或L集群内R小于1/9、同时H集群内R大于9的位点,即为关联位点。
1.5 候选基因的挖掘及其功能注释
根据集群分离分析结果,以连续存在3个以上关联位点的区域为强关联区,并将强关联区内所包含的基因作为候选基因,最后根据大豆基因组注释信息对候选基因进行功能注释。
2 结果与分析
2.1 大豆HapMap数据预处理及点突变比率的计算
经预处理,最终可以用于分析的大豆HapMap数据包含15 391份大豆种质在42 449个位点上的分型结果。由于突变发生的概率很低,故将每个位点上基因型比例小于50%的位点作为突变点。经统计,大豆种质间点突变比率R_snp为0.089~0.531,平均变异率为0.261;但个体间变异率差异很大,变异系数为27.3%,且呈混合分布模式(图1)。
2.2 大豆种质突变热点区数目的确定
经计算,用于分析的15 391份大豆种质中95%的突变热点间距不超过54 784.6 bp(图2),故本研究的窗口大小确定为Wlength=54 800 bp,步长为2 740 bp。
相邻两位点间距的平均值(μlen)为 11 092 bp,则一个窗口范围内所包含的点突变数目的期望值Esnp=Wlength/μlen=4.94≈5个,说明突变热点判定阈值Thotspot的取值范围为大于5的一切自然数。本研究拟通过点突变比率和突变热点区数目2个表型来探讨突变热点的产生机制,为了尽量消除点突变比率对突变热点区数目的影响,以满足2个表型间相关系数>0.5的最大自然数为最终的N值。经计算,最终的阈值Thotspot=13,此时点突变比率与突变热点区数目2个表型间的相关系数为0.53。根据上述参数统计每份大豆种质的突变热点数分布情况,以大豆种质PI416792染色体Gm01为例,其突变热点数如图3所示。

图 1 15 391份大豆种质点突变比率的分布
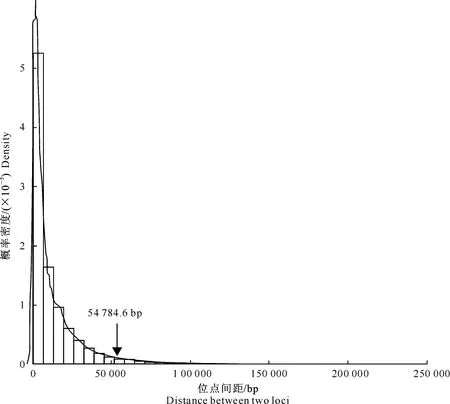
图 2 15 391份大豆种质突变热点间距的分布
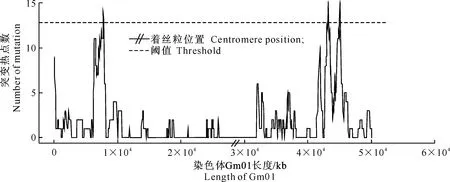
图 3 大豆种质PI416792染色体Gm01的突变热点分布
Fig.3 Number of mutation hotspots of PI416792 on Gm01
经统计,15 391份大豆种质的突变热点区数目为5~1 324个,平均每份种质包含347.8个突变热点区。个体间的变异程度远大于点突变比率,其变异系数为62.4%,但也呈混合分布模式(图4)。

图 4 15 391份大豆种质突变热点区数目分布
Fig.4 Number distribution of mutation hotspots among 15 391 soybean accessions
2.3 大豆种质点突变比率及突变热点区数目的关联位点挖掘
2.3.1 混合群体分群 如图1和图4所示,15 391份大豆种质的点突变比率及突变热点区数目均呈混合分布模式。为了能客观地分群,采用EM算法进行GMM参数估计。通过参数估计,可将点突变比率及突变热点区数目2个表型分为高均值群体和低均值群体(图5)。以突变热点区数目为例,其分布可看作由2个高斯分布组成,根据2个峰值的位置及群体标准差,初始值设置为:均值μ=(200,400),标准差δ=(100,200),权重α=(0.3,0.7);经过36轮循环,各参数趋于稳定,将初始值最终修正为:μ=(211.8,485.9),δ=(91.7,220.1),α=(0.50,0.50)。采用同样的方法,确定组成点突变比率分布的2个高斯分布参数为:μ=(0.197,0.260),δ=(0.029,0.067),α=(0.52,0.48)。
最后,根据GMM参数估计结果进行分群,经计算,突变热点区数目<160的大豆种质有超过90%来自低均值群体(μ=211.8),而突变热点区数目为340~720的大豆种质有超过90%的来自于高均值群体(μ=485.9)。根据同样的判定方法,确定点突变比率<0.193的大豆种质有超过90%的个体来自低均值群体(μ=0.197),而点突变比率>0.247的大豆种质有超过90%的个体来自于高均值群体(μ=0.260)。

图 5 基于GMM参数估计结果的大豆种质突变热点区数目(a)及点突变比率(b)的群体分布
2.3.2 关联位点的确定 根据上述分群结果,分别统计2种表型的低均值和高均值群体内2种基因型‘1’和‘0’的比值,进行标记位点与表型的关联性分析。以连续存在3个以上关联位点的区间为强关联区,最终在Gm16、Gm17和Gm19上各获得1个与点突变率存在强关联的区间,范围分别为29 144 428-30 604 603,12 107 463-12 147 725和45 000 827-45 178 132 bp;在Gm16和Gm17上各获得1个与突变热点区数目存在强关联的区间,范围为29 153 474-20 604 603和12 133 293-12 210 561 bp(表1)。

表 1 大豆种质点突变比率及突变热点区数目的强关联区标记信息汇总
注:*表示不满足判定条件标记的关联系数,N表示不相关,Y表示相关。
Note:* indicates the non-compliance association coefficient,N denotes no and Y denotes yes.
2.4 大豆种质增变候选基因的挖掘及其功能注释
2.4.1 候选基因的挖掘 根据关联位点的挖掘结果,以大豆种质点突变比率和突变热点区数目2个表型同时定位到的区间,即二者结果的交集进行获选基因的挖掘。结果显示,Gm16上的29 153 474-30 604 603 bp和Gm17上的12 133 293-12 147 725 bp片段所包含的编码序列为候选基因。其中,Gm16上包含115个候选基因(Glyma16g25185.1~Glyma16g26500.1);Gm17上包含103个候选基因(Glyma17g15380.1~Glyma17g15490.1)。由于篇幅有限,本研究只列出部分候选基因,结果见表2。
2.4.2 候选基因的功能注释 参考大豆基因组Wm82.a2.v1所包含的信息对上述基因进行功能注释,结果见表2。由表2可见,Gm16上的Glyma16g26440.1和Gm17上的Glyma17g15420.1同属于nudix水解酶基因家族,Gm16上的Glyma16g25330.1和Gm17上的Glyma17g15530.1同属于α/β-水解酶家族,其他基因都仅在其中一个染色体区间存在。进一步的功能分析表明,nudix水解酶家族基因编码蛋白为具有不同程度底物特异性的水解酶,可以水解包括核苷二磷酸、核苷三磷酸及RNA帽等一系列有机焦磷酸盐[25]。这些功能均与基因组突变有关,因此将Glyma16g26440.1和Glyma17g15420.1作为大豆基因组候选增变基因。

表 2 大豆种质部分增变候选基因及其功能注释信息
注:加粗字体表示在2条染色体上同时挖掘到的属于同一基因家族的基因。
Note:The bold font indicates the genes identified on both chromosomes and belong to the same gene family.
3 讨 论
3.1 研究大豆增变基因的意义
随着很多植物全基因组测序的完成,世界上许多实验室迅速展开了大规模建立突变体库的工作,这直接促进了植物基因功能研究及作物性状改良的发展[26-27]。植物的自发突变率很低,通过化学手段、物理手段可以有效提高其突变率,但仍然需要构建庞大的突变体库来进行突变体的筛选。例如,赵永亮等[28]利用EMS处理玉米自交系黄早4和7922的成熟花粉,在全部2 754个EMS诱变后代M2果穗中,发现白色胚乳基因y1的突变体有97个[28]。通过研究增变基因来提高植物突变率,将大大促进作物突变育种的发展,特别是像大豆这种异花授粉难度很大的作物。
3.2 改进的超级集群分离分析法的优点
为了更好地进行数据挖掘,本研究在Super-BSA方法的基础上进行了如下改进。第一,为了准确描述突变热点这一表型,采用滑动窗口法来定义突变热点,同时采用突变热点区数目来反映某个体突变程度的高低。所谓的突变热点区数目,即在基因组上以单位步长移动窗口,满足一个窗口内所包含的突变点数目超过阈值的窗口的数目,这一数值是突变热点区数目及突变热点区长度平衡后的结果,可以客观地反映每个个体的突变情况;第二,在进行阈值确定时,以满足突变热点区数目与点突变比率相关系数大于0.5的最大自然数为选择标准,目的是降低点突变比率对突变热点区数目的干扰,从而更准确地进行基因定位;第三,与其他研究直接选择群体两端相同数目个体进行分群[22]不同的是,本研究首先采用EM算法来估计组成混合分布中的每种高斯分布,再根据混合分布中某类个体所占的比率进行分群。这样不仅可以做到客观分群,还可为下一步的关联位点分析判定标准的选择提供参考。本研究所采用的改进的Super-BSA方法也可以扩展到其他物种的不同性状研究中。
3.3 大豆增变候选基因的功能
本研究利用大豆基因组Wm82.a2.v1及HapMap数据,采用Super-BSA鉴定出2个同属于nudix水解酶基因家族的基因Glyma16g26440.1和Glyma17g15420.1。研究表明,在大肠杆菌中,nudix的直系同源基因MutT具有抑制基因突变的功能[29]。而植物中,由于nudix水解酶的底物特异性,其功能已经延伸到细胞响应调控的各个方面[30-32],但植物nudix水解酶基因与基因突变的关系尚不明确,而本研究的结果将为这一问题的阐明提供理论依据。
4 结 论
本研究发现,不同的大豆种质的点突变比率及突变热点区数目存在较大差异,如点突变比率为0.089~0.531,突变热点区数目为5~1 324个。因此可以将点突变比率和突变热点区数目作为表型用于分析与大豆突变相关的基因,且这2个表型皆符合GMM模型,通过参数估计,可分别将以上2个表型分为高均值群体和低均值群体,此分群结果可作为Super-BSA的分群依据。通过位点与性状的关联性分析,分别在Gm16和Gm17上定位到与2个表型同时存在强关联性的位点,共包含218个候选基因,其中,Gm16上的Glyma16g26440.1和Gm17上的Glyma17g15420.1同属于nudix水解酶基因家族,可作为大豆基因组候选增变基因进行进一步的研究。
[1]Aminetzach Y T,Macpherson J M,Petrov D A.Pesticide resistance via transposition-mediated adaptive gene truncation inDrosophila[J].Science,2005,309(5735):764-767.
[2]Burrus V,Waldor M K.Shaping bacterial genomes with integrative and conjugative elements [J].Res Microbiol,2004,155(5):376-386.
[3]Benzer S.On the topography of the genetic fine structure [J].Proc Natl Acad Sci USA,1961,47(3):403-415.
[4]Suckow J,Markiewicz P,Kleina L G,et al.Genetic studies of the Lac repressor:ⅩⅤ.4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure [J].J Mol Biol,1996,261(4):509-523.
[5]Betz A G,Neuberger M S,Milstein C.Discriminating intrinsic and antigen-selected mutational hotspots in immunoglobulin V genes [J].Immunol Today,1993,14(8):405-411.
[6]陆思千,贾舒婷,罗 瑛.突变p53功能研究新进展与个性化的肿瘤治疗新策略 [J].遗传,2011,33(6):539-548.
Lu S Q,Jia S T,Luo Y.Recent advances in mutant p53 and novel personalized strategies for cancer therapy [J].Hereditas,2011,33(6):539-548.
[7]Krawczak M,Smith-Sorensen B,Schmidtke J,et al.Somatic spectrum of cancer-associated single basepair substitutions in theTP53 gene is determined mainly by endogenous mechanisms of mutation and by selection [J].Hum Mutat,1995,5(1):48-57.
[8]Lee K J,Hwang J E,Velusamy V,et al.Selection and molecular characterization of a lipoxygenase-free soybean mutant line induced by gamma irradiation [J].Theor Appl Genet,2014,127(11):2405-2413.
[9]Song Q,Hyten D L,Jia G,et al.Development and evaluation of SoySNP50K,a high-density genotyping array for soybean [J].PLoS One,2013,8(1):e54985.
[10]Cooper D N,Youssoufian H.The CpG dinucleotide and human genetic disease [J].Hum Genet,1988,78(2):151-155.
[11]Krawczak M,Ball E V,Cooper D N.Neighboring nucleotide effects on the rates of germ-line single-base-pair substitution in human genes [J].Am J Hum Genet,1998,63(2):474-488.
[12]Isono K,Yourno J.Chemical carcinogens as frameshift mutagens:SalmonellaDNA sequence sensitive to mutagenesis by polycyclic carcinogens [J].Proc Natl Acad Sci U S A,1974,71(5):1612-1617.
[13]Ehrlich S D,Bierne H,d’Alencon E,et al.Mechanisms of illegitimate recombination [J].Gene,1993,135(1/2):161-166.
[14]Tran H T,Degtyareva N P,Koloteva N N,et al.Replication slippage between distant short repeats inSaccharomycescerevisiaedepends on the direction of replication and theRAD50 andRAD52 genes [J].Mol Cell Biol,1995,15(10):5607-5617.
[15]You Y H,Li C,Pfeifer G P.Involvement of 5-methylcytosine in sunlight-induced mutagenesis [J].J Mol Biol,1999,293(3):493-503.
[16]Rogozin I B,Pavlov Y I.Theoretical analysis of mutation hotspots and their DNA sequence context specificity [J].Mutat Res,2003,544(1):65-85.
[17]郑乃刚,高涵昌,吴景兰,等.野生型DNA聚合酶β转染Eca-109细胞后增变基因表型变化 [J].郑州大学学报(医学版),2008,43(6):1085-1090.
Zheng N G,Gao H C,Wu J L,et al.Changes of mutator phenotypes in Eca-109 cells transfected with wild type DNA polymerase beta [J].Journal of Zhengzhou University (Medical Sciences),2008,43(6):1085-1090.
[18]Roberts S A,Lawrence M S,Klimczak L J,et al.An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers [J].Nat Genet,2013,45(9):970-976.
[19]Jonathan G,Avraham A L.Stress,mutators,mutations and stress resistance [M]//Pareek A,Sopory S K,Bohnert H,et al.Abiotic stress adaptation in plants. [s.l.]:Springer Netherlands,2010:471-483.
[20]Hoffman P D,Leonard J M,Lindberg G E,et al.Rapid accumulation of mutations during seed-to-seed propagation of mismatch-repair-defectiveArabidopsis[J].Genes Dev,2004,18(21):2676-2685.
[21]廖 毅,孙保娟,孙光闻,等.集群分离分析法在作物分子标记研究中的应用及问题分析 [J].分子植物育种,2009,7(1):162-168.
Liao Y,Sun B J,Sun G W,et al.The application and key problems of bulked segregant analysis on the research of molecular marker in crop [J].Molecular Plant Breeding,2009,7(1):162-168.
[22]Takagi H,Abe A,Yoshida K,et al.QTL-seq:rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations [J].Plant J,2013,74(1):174-183.
[23]Schmutz J,Cannon S B,Schlueter J,et al.Genome sequence of the palaeopolyploid soybean [J].Nature,2010,463(7278):178-183.
[24]Purcell S,Neale B,Todd-Brown K,et al.PLINK:a tool set for whole-genome association and population-based linkage analyses [J].Am J Hum Genet,2007,81(3):559-575.
[25]McLennan A G.The nudix hydrolase superfamily [J].Cell Mol Life Sci,2006,63(2):123-143.
[26]张帆涛,方 军,孙昌辉,等.水稻矮秆突变体dtl1的分离鉴定及其突变基因的精细定位 [J].遗传,2012,34(1):81-88.
Zhang F T,Fang J,Sun C H,et al.Characterisation of a rice dwarf and twist leaf 1 (dtl1) mutant and fine mapping ofDTL1 gene [J].Hereditas,2012,34(1):81-88.
[27]徐建龙,王俊敏,骆荣挺,等.空间诱变水稻大粒型突变体的遗传育种研究 [J].遗传,2002,24(4):431-433.
Xu J L,Wang J M,Luo R T,et al.Studies of inheritance and application in rice breeding of large grain mutant induced in space environment [J].Hereditas,2002,24(4):431-433.
[28]赵永亮,宋同明,马惠平.利用花粉化学诱变快速创造特用玉米新种质 [J].作物学报,1999,24(2):157-161.
Zhang Y L,Song T M,Ma H P.The quick development of specialty corn by chemical mutagenesis of pollen [J].Acta Agronomica Sinica,1999,24(2):157-161.
[29]Nakabeppu Y,Sakumi K,Sakamoto K,et al.Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids [J].Biol Chem,2006,387(4):373-379.
[30]Yoshimura K,Shigeoka S.Versatile physiological functions of the Nudix hydrolase family inArabidopsis[J].Biosci Biotechnol Biochem,2015,79(3):354-366.
[31]Muthuramalingam M,Zeng X,Iyer N J,et al.A GCC-box motif in the promoter of nudix hydrolase 7 (AtNUDT7) gene plays a role in ozone response ofArabidopsisecotypes[J].Genomics,2015,105(1):31-38.
[32]Fonseca J P,Dong X.Functional characterization of a Nudix hydrolase AtNUDX8 upon pathogen attack indicates a positive role in plant immune responses [J].PLoS One,2014,9(12):e114119.
Identification of mutators in soybean genome by super bulked segregant analysis
CHANG Wei,WANG Juan,HUANG Zhigang,CHEN Jibao
(College of Agricultural Engineering,Nanyang Normal University,Nanyang,Henan 473061,China)
【Objective】 In this study,the soybean genome Wm82.a2.v1 and HapMap were used to identify the potential mutators.【Method】 After pretreatment,the soybean HapMap data (comprising 52 041 genotype from 19 652 soybean germplasm) were used for association loci mining by improved Super-BSA method with selected size of the sliding window,step size and threshold parameters.The candidate genes associated with both point mutation rate and the number of mutation hotspots were considered as soybean mutators.All these genes are used for functional prediction by the soybean genome Wm82.a2.v1.【Result】 After the pretrtatment of HapMap data,15 391 individuals were retained.Among these,the range of mutation rate was 0.089-0.531 with an average mutation rate of 0.261,and the range of number of mutation hotspots was 5-1 324 with a mean of 347.8.Super bulked segregant analysis also showed that the intervals 29 153 474-30 604 603 bp on Gm16 and 12 133 293-12 147 725 bp on Gm17 were two regions with strong associations with both phenotypes.Glyma16g26440.1 on Gm16 andGlyma17g15420.1 on Gm17 belonged to the nudix hydrolase superfamily involving in genome mutations.【Conclusion】 The two members of nudix hydrolase (Glyma16g26440.1 andGlyma17g15420.1) were associated with mutation rate and the number of mutation hotspots,and could be considered as candidate soybean mutators for further studies.
soybean;mutation hotspots;mutators;Super-BSA;nudix hydrolase genes
时间:2016-10-20 16:36
10.13207/j.cnki.jnwafu.2016.12.010
2015-07-10
南阳师范学院科研基金项目(ZX2015012)
常 玮(1984-),男,黑龙江齐齐哈尔人,讲师,博士,主要从事大豆分子育种研究。
陈吉宝(1969-),男,河南南阳人,副教授,硕士生导师,主要从事作物种质资源研究。
Q319.32;S565.1
A
1671-9387(2016)12-0064-09
网络出版地址:http://www.cnki.net/kcms/detail/61.1390.S.20161020.1636.020.html
猜你喜欢
中学化学(2024年4期)2024-04-29 22:54:35
数学物理学报(2022年1期)2022-03-16 06:15:20
现代园艺(2017年21期)2018-01-03 06:41:32
中国民族医药杂志(2016年5期)2016-05-09 07:43:50
作文大王·低年级(2016年3期)2016-03-11 00:48:53
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
中国惯性技术学报(2015年1期)2015-12-19 13:12:07
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
海南医学(2010年17期)2010-03-21 07:43:18
