大空间位阻β-二亚胺镁配合物的合成、晶体结构及硅氢化反应
2016-12-05 11:49:28马猛涛沈兴超于志娟姚薇薇杜丽婷徐莉
无机化学学报 2016年10期
马猛涛 沈兴超 于志娟 姚薇薇 杜丽婷 徐莉
(1南京林业大学理学院,南京210037)
(2南京中医药大学药学院,南京210023)
大空间位阻β-二亚胺镁配合物的合成、晶体结构及硅氢化反应
马猛涛*,1沈兴超1于志娟1姚薇薇2杜丽婷1徐莉*,1
(1南京林业大学理学院,南京210037)
(2南京中医药大学药学院,南京210023)
2,4-戊二酮分别与不同的伯胺反应,合成出2种新的大空间位阻的β-二亚胺配体。其中β-二亚胺配体1与格氏试剂甲基碘化镁(MeMgI)反应得到相应四配位镁的甲基配合物,配体1的锂盐与溴化镁(MgBr2)反应制备出Mg-Li双金属溴配合物。新β-二亚胺配体和相应镁的甲基配合物和溴配合物的晶体结构均通过单晶X射线衍射确定,相应镁的甲基配合物和溴配合物在苯乙酮的硅氢化反应中显示了较好的催化活性。
镁;β-二亚胺配体;晶体结构;硅氢化
0 Introduction
Due to its ease of synthesis and the fact that the substituents on the aromatic amine and backbone are readily tuned,β-diketiminate ligand is a versatile precursor which iswidely used in inorganic chemistry, organic chemistry and catalysis.It can be used for the coordination of almost allmain and transition metals, and often used as a very efficient catalyst for olefin polymerization and in organic catalysis[1-3].As early as 1968 there has been a report aboutβ-diketiminate ligand as single electron bidentate ligand to prepare
nickel(II)complexes[4].Recently,Shen et al.used it to synthesize a series of rare earth complexes which are highly efficient catalysts for the polymerization of L-lactide[5].
Over the past decade,various kinds of ligands stabilizing subvalent metal complexes have been synthesized.For example,Robinson et al.have successfully synthesized monovalent zinc complex [(DippNacnac)Zn(II)]2with the correspondingβdiketiminate ligand Dipp Nacnac(DippNacnac=(Dipp NC(Me))2CH,Dipp=2,6-diisopropylphenyl)in 2005[6]. Subsequently Jones et al.also used the same ligand Dipp Nacnac to synthesize the first room temperature stable monovalent magnesium compound[(Dipp Nacnac)Mg(II)]2in 2007 and further extended it to other similarβ-diketiminate ligands[7-8].These Mg(II)complexes could be used as a reducing agent in organic,organometallic and inorganic reactions[9-11],in some ways it can even replace the traditional reducing agents used in many synthesis reactions.For instance, Driess and Power et al.respectively prepared cyclic and acyclic carbene silicon analogues with[(Mes Nacnac)Mg(II)]2as a reducing agent recently[12-13]. However,most of the known ligands cannot stabilize other active low valence metal complexes such as calcium and rare earth because of the comparatively slow steric hindrance.Moreover the reports aboutβdiketiminate magnesium complexes are relatively few. Herein we report the syntheses of two new extremely bulkyβ-diketiminate ligands,the correspondingβdiketiminate magnesium methyl and Mg-Li bimetallic complexes and their application in the catalytic hydrosilylation of acetophenone.
1 Experimental
1H and13C{1H}NMR spectra were recorded on Bruker AvanceⅢ600 MHz spectrometer and were referenced to the resonances of the solvent used. HRMS data was obtained with the Thermo Scientific LTQ Orbitrap XL.Melting pointswere determined and uncorrected.The starting material 2,6-bis (diphenylmethyl)-4-isopropylaniline and 2,6-bis (dinaphthylmethyl)-4-methylaniline were prepared according to literatures[14-15].Other chemicals were purchased and used without further purification.
1.1 Synthesis of compound 1
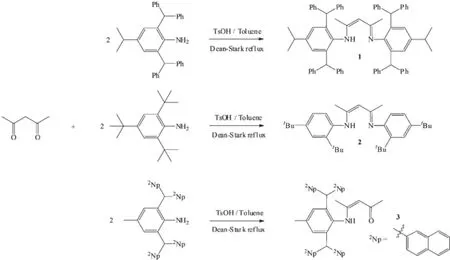
2,4-Pentanedione(0.20 mL,2.24 mmol)and 2,6-bis(diphenylmethyl)-4-isopropylaniline(2.00 g,4.28 mmol)were refluxed with p-toluenesulfonic acid(0.83 g,4.36 mmol)in toluene(80 mL)under Dean-Stark conditions for 3 days.Upon cooling of the resulting yellow mixture,a beige solid was precipitated,which was filtered,dissolved in 100 mL CH2Cl2,neutralized with 70mL of a 5%(w/w)aqueous NaOH solution,and extracted into CH2Cl2(2×50m L).After dried by MgSO4,the solvent was removed in vacuo and compound 1 was recrystallized from CH2Cl2/hexane as colorless crystals(Yield:65%).m.p.253~254℃.1H NMR(600 MHz,298 K,CDCl3):δ12.01(s,1H,N H), 7.23~6.97(m,40H,Ar-H),6.86(s,4H,Ar-H),5.90 (s,4H,C H Ph2),4.12(s,1H,=C H),2.74(sep,3JHH=7.2 Hz,2H,C H(CH3)2),1.08(d,3JHH=7.2 Hz,12H,CH (C H3)2),0.19(s,6H,NCCH3).13C{1H}NMR(151 MHz, 298 K,CDCl3)δ164.0(C=N),144.9,144.3,142.7, 141.6,138.5,130.0,129.4,128.2,128.0,126.7, 126.1,125.8(Ar-C),94.7(=C H),52.3(C HPh2),33.6 (C H3),24.0(CH(C H3)2),19.8(C H(CH3)2).HRMS(ESI): m/z Calcd.for C75H70N2+H[M++H]:999.5618;Found: 999.5667.
1.2 Synthesis of com pound 2
Compound 2 was prepared using a similar procedure to that employed for 1(Yield:44%).m.p. 202~203℃.1H NMR(600 MHz,298 K,CDCl3):δ 12.31(s,1H,N H),7.33(s,2H,Ar-H),7.11(d,3JHH= 8.4Hz,2H,Ar-H),6.70(d,3JHH=8.4 Hz,2H,Ar-H), 4.87(s,1H,=C H),1.81(s,6H,NCC H3),1.31(s,18H, C(C H3)3),1.30(s,18H,C(C H3)3).13C{1H}NMR(151 MHz,298 K,CDCl3)δ159.5(C=N),146.3,142.2, 142.0,125.8,123.1,122.7(Ar-C),96.4(NC=C H), 35.3,34.5(C(CH3)3),31.6,30.6(C(C H3)3),21.3(C H3). HRMS(ESI):m/z Calcd.for C30H50N2+H[M++H]: 475.4052;Found:475.4034.
1.3 Synthesis of compound 3
Compound 3 was prepared using a similar procedure to that employed for 1(Yield:67%).m.p. 157~158℃.1H NMR(600 MHz,298 K,CDCl3):δ
12.20(s,1H,N H),7.81~7.28(m,28H,Ar-H),6.91(s, 2H,Ar-H),5.95(s,2H,C H(2Np)2),4.90(s,1H,=C H), 2.19(s,3H,C H3),2.07(s,3H,C H3),0.69(s,3H, C H3).13C{1H}NMR(151 MHz,298 K,CDCl3)δ195.6 (C=O),163.9(C=N),142.5,140.8,139.4,137.4, 134.3,133.44,133.41,132.3,132.2,129.8,128.5, 128.1,128.01,127.97,127.88,127.82,127.60, 127.58,126.1,125.9,125.8,125.7(Ar-C),96.3 (=CH),52.4(C H(2Np)2),29.0,21.7,18.2(C H3).HRMS (ESI):m/z Calcd.for C54H43NO+H[M++H]:722.3424; Found:722.3463.
1.4 Synthesis ofm agnesium methyl complex 4
MeMgI(0.67mL,2.01mmol,3 mol·L-1in Et2O) was added dropwise to a solution of ligand 1(1.00 g, 1.00 mmol)in THF at-70℃.The mixture was warmed to room temperature and stirred overnight, filtered and concentrated.Hexane was added to give colorless crystals of 4(0.79 g,Yield:72%).m.p. 146.8~148.2℃.1H NMR(600 MHz,298 K,C6D6):δ 7.38~6.86(m,44H,Ar-H),6.13(s,4H,C H Ph2),4.60 (s,1H,=C H),3.58(m,4H,THF),2.50(sep,3JHH=7.2 Hz,2H,C H(CH3)2),1.34(m,4H,THF),0.97(s,6H, NCC H3),0.96(d,3JHH=7.2 Hz,12H,C H(CH3)2),-0.81 (s,3H,MgC H3).13C{1H}NMR(151 MHz,298 K, C6D6):δ170.8(C=N),146.3,146.0,144.0,142.9, 138.8,130.4,129.7,128.3,128.0,126.3,125.9(Ar-C),95.9(=C H),67.9(THF),51.6(C HPh2),33.3(C H3), 25.3(THF),24.1(C H(CH3)2),23.6(C H(CH3)2),-15.1 (MgCH3).
1.5 Synthesis ofm agnesium brom ide complex 5
nBuLi(0.7 mL,1.12 mmol,1.6 mol·L-1in hexane)was added dropwise to a solution of ligand 1 (0.97 g,0.97 mmol)in THF at room temperature and stirred for 2 h.The resulting red-brown solution was added to MgBr2(0.18 g,0.97 mmol)in THF and stirred overnight.The solventswere removed in vacuo and the residue was extracted with toluene and filtered.Hexane was added to afford the colorless crystals of 5(0.73 g,Yield:56%).m.p.193.5~195.6℃.1H NMR(600 MHz,298 K,C6D6):δ7.52~6.94(m, 44H,Ar-H),6.22(s,4H,C H Ph2),4.56(s,1H,=C H), 2.63(sep,3JHH=7.2 Hz,2H,CH(C H3)2),1.06(d,3JHH= 7.2 Hz,12H,C H(CH3)2),0.89(s,6H,NCC H3).13C{1H} NMR(151MHz,298 K,C6D6):δ166.5(C=N),150.6, 146.0,144.4,141.9,137.6,130.4,130.0,128.0,127.9, 126.6,126.3,126.1(Ar-C),93.9(=C H),52.6(C HPh2), 33.8(C H3),24.3(CH(C H3)2),23.2(C H(CH3)2).
1.6 X-ray crystal structure determ ination
Diffraction data were collected on a Bruker D8 VENTURE PHOTON 100 diffractometer with Mo Kα radiation(λ=0.071 073 nm,graphitemonochromator)at 135(2)K.SADABS absorption corrections were applied[16].The structureswere solved by directmethods and refined on F2by full matrix least squares (SHELXL-97)using all unique data[17].All nonhydrogen atoms were refined anisotropically,while the hydrogen atomswere introduced at calculated positions and refined riding on their carrier atoms.Crystal data for the complexes 1,2,4,5 and a summary of the crystallographic analysesweregiven in Table1.
CCDC:1445844,1;1445845,2;1480136,4; 1480137,5.
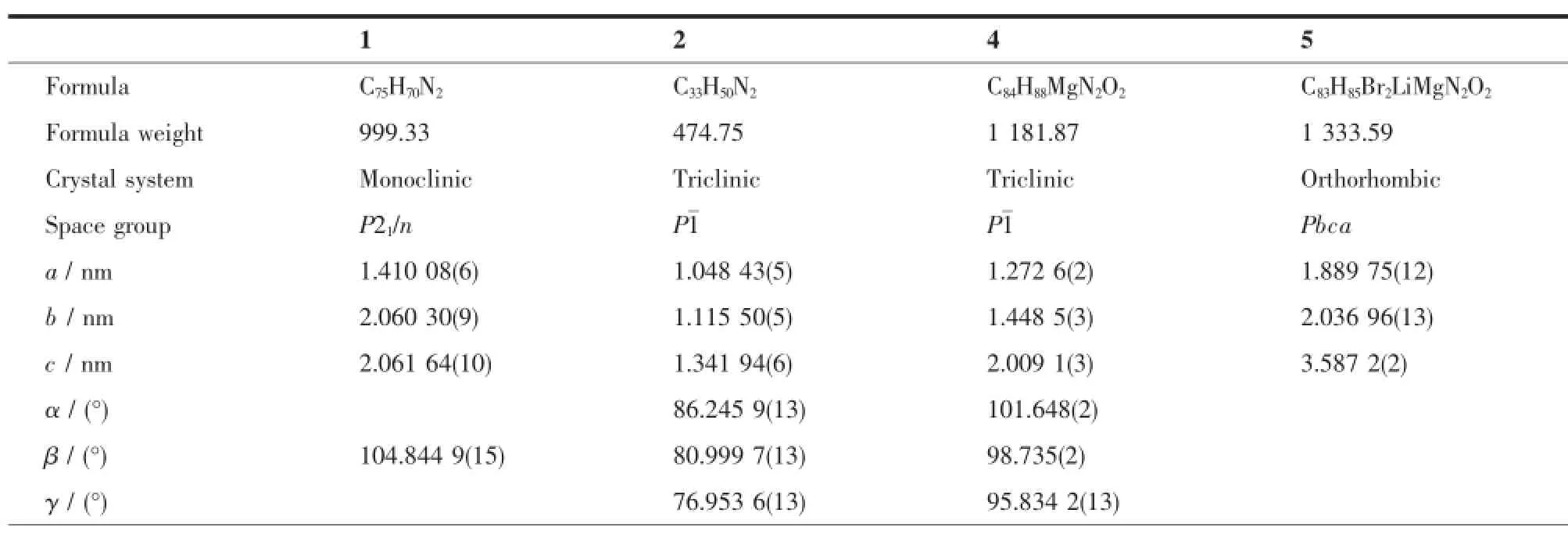
Tab le 1 Crystallographic data for 1,2,4 and 5
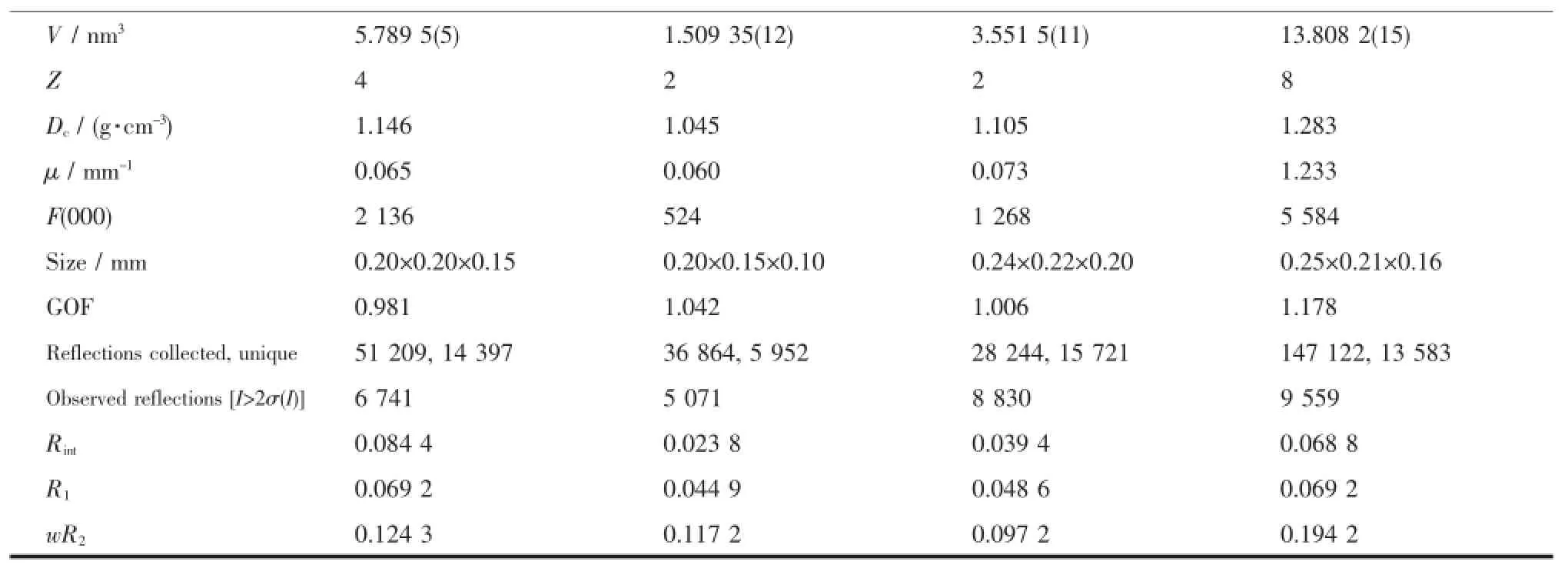
Continued Table 1
2 Results and discussion
When 2,6-bis(diphenylmethyl)-4-isopropylaniline and 2,4-pentanedione were refluxed in ethanol or toluene as per protocol followed in normalβdiketiminate ligand synthesis,no reaction wasobserved even when the longer reaction time(2 weeks)was employed.However 2,4-pentanedione upon treatment with 2 equiv.of 2,6-bis(diphenylmethyl)-4-isopropylaniline and p-toluenesulfonic acid by Dean-Stark reflux for 3 days in toluene afforded the correspondingβdiketiminate ligand 1 in good yield(65%)(Scheme 1). Nuclear magnetic resonance(NMR)spectroscopic analysis revealed that in the1H NMR spectrum(CDCl3) the NH2signal(δ3.26)of the starting material 2,6-bis(diphenylmethyl)-4-isopropylaniline[14]disappeared while a new singletsignalappeared atδ12.01 which is assigned to the NH characteristic resonance and another new singlet signal atδ4.12 that is assigned to the backbonemethine resonance of compound 1.Other integration data and chemical shifts for all other signals are consistent with the desired target product 1.The13C{1H}NMR spectrum of compound 1 further confirmed the structure.It displayed a characteristic downfield C=N resonance atδ164.0 and C=C double bond resonance atδ94.7.Finally the solid state molecular structure of ligand 1 was determined by single-crystalX-ray diffraction.
Colorless crystals of 1 suitable for X-ray diffraction study were obtained from mixed dichloromethane and hexane solvents(Fig.1).Selected bond lengths and angles are listed in Table 2.In
compound 1,the double bond C(38)-C(39)(0.1380(3) nm)is shorter than the single bond C(36)-C(38) (0.142 3(3)nm),the bond length of C-N single bond C (39)-N(2)(0.134 6(2)nm)is slightly longer than that of the C=N double bond C(36)-N(1)(0.131 5(3)nm), the bond distances of aromatic N(1)-C(1)and N(2)-C(41)are almost identical(0.142 6(2)and 0.142 7(2) nm)and aremuch longer than that of C(39)-N(2)and slightly longer than that of the reported mesityl substitutedβ-diketiminate ligand(0.142 4(2)nm)[18]. This could be attributed to the steric hindrance of the bulky 2,6-bis(diphenylmethyl)-4-isopropylaniline.The N-H hydrogen was fixed to the nitrogen with the longer C-N bond.The N(1),C(36),C(38),C(39)and N (2)atoms are almost in the same plane.Their bond angles are around the ideal value of 120°.
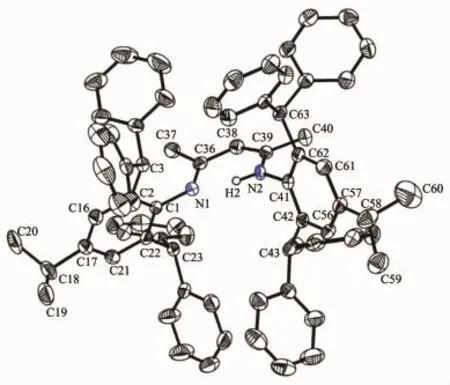
Fig.1 Molecular structure of 1 with thermal ellipsoids at the 50%probability level
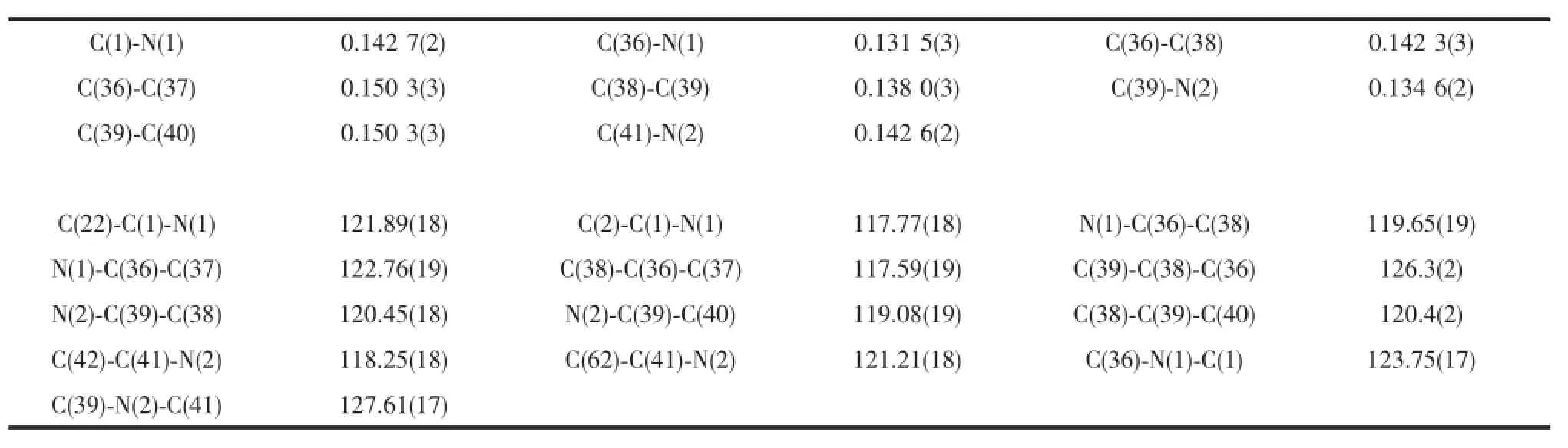
Table 2 Selected bond lengths(nm)and angles(°)for com pound 1
When the similar reaction between 2,4-pentanedione and 2,4,6-tri-tert-butyl aniline was carried out under Dean-Stack reflux in toluene for 3 days,a new product was produced(Scheme 1).This product also exhibited a NH characteristic resonance atδ12.31 and a backbone methine resonance atδ 4.87 in the1H NMR spectrum(CDCl3)as well as a characteristic C=N double bond resonance atδ159.5 and C=C double bond resonance atδ96.4 in the13C {1H}NMR spectrum(CDCl3).However,integration indicated that there were 36 protons atδ1.31 and 1.30.This is notmatched with the expected product in which six tertiary butyl groups should have 54 protons.Meanwhile there are six aromatic protons which are twomore than the expected product.Hence we speculated that two tertiary butyl groups of phenyl
ring could be lost.The high resolution mass spectrum (HRMS)also demonstrated that the experimental observed value of 475.403 4 is same to the calculated value of 475.405 2.
However,from the above1H,13C{1H}NMR and HRMS spectra,we did not know which tertiary butyl groupswere lost.We attempted to grow single crystals of this new compound.The single-crystal X-ray diffraction analysis of 2 established that the two orthotertiary butyl groups of phenyl ring were indeed lost. We deduced the high reaction temperature and time maybe lead to the tertiary butyl group decomposition. We subsequently lowered the reaction temperature from reflux in toluene to room temperature,however no reaction was observed even after two weeks.Selected bond lengthsand anglesare listed in Table 2.Asshown in Fig.2,one ortho-substituted tertiary butyl group of each phenyl ring was obviously split from the original 2,4,6-tri-tert-butylaniline.The bond lengths of C(6)-N (1)and C(10)-N(2)(0.142 36(15)and 0.141 94(16)nm) are slightly shorter than those of 1 and other similarβdiketiminate ligands reported previously[18-19]due to the vacancy generated by the cleaved ortho-tertiary butyl groups as expected.Moreover the two ortho-tertiary butyl groups adopt a trans-arrangement in order to minimize steric crowding.
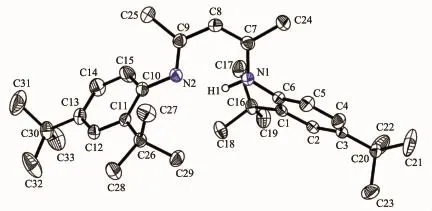
Fig.2 Molecular structure of 2 with thermal ellipsoids at the 50%probability level
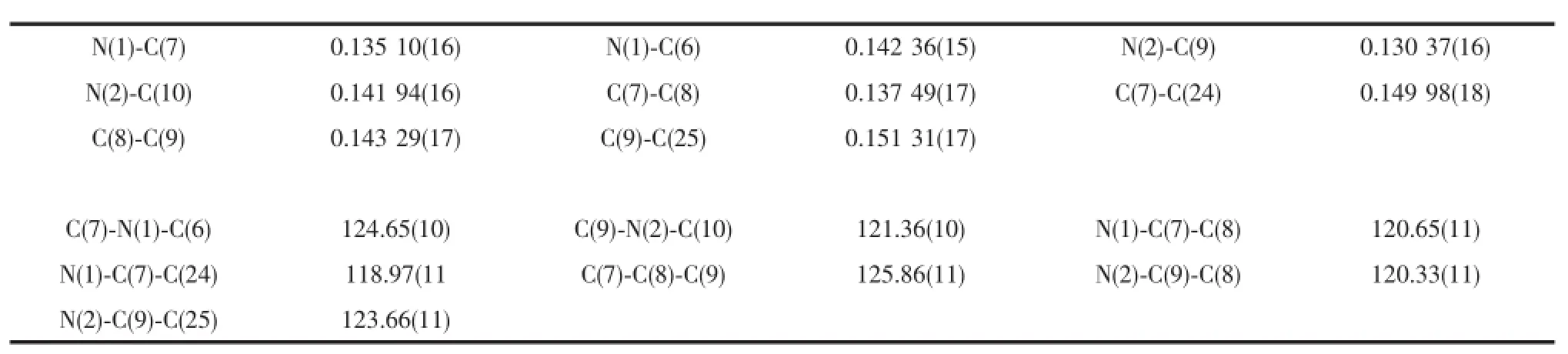
Tab le 3 Selected bond lengths(nm)and angles(°)for com pound 2
Quite recently,the extremely bulky 2,6-bis (dinaphthylmethyl)-4-methylaniline was synthesized via a two step Grignard reaction and Friedel-Crafts alkylation protocol in excellent yields and successfully applied to prepare what can be categorized as one of the bulkiest N-heterocyclic carbenes synthesized so far[15].Hence we envisaged that we can prepare the correspondingβ-diketiminate ligand.When 2,6-bis (dinaphthylmethyl)-4-methylaniline was treated with 2,4-pentanedione under the same reaction condition as above,only compound 3 was obtained as observed via1H and13C{1H}NMR spectroscopic analysis(Scheme 1).There is a NH characteristic resonance atδ12.20 and a backbone methine resonance atδ4.90 in the1H NMR spectrum(CDCl3)aswell as a characteristic unreacted C=O double bond resonance atδ195.6 and the new C=C double bond resonance atδ96.3 in the13C{1H}NMR spectrum(CDCl3).We suppose the first
equivalent primary amine underwent reaction and the second did not further reactwith another ketone group of 2,4-pentanedione because of much larger steric hindrance.We also extended the reaction time from 5 days to 10 days,the same product was obtained. Compared with the above-mentioned two reactions 2,6-bis(dinaphthylmethyl)-4-methylaniline should be the bulkiest primary amine reported to date.
The reaction of ligand 1 with one equiv.MeMgIin THF did not give the desired pure magnesium iodide complex.1H NMR analysis indicated that it could generate the magnesium iodide,magnesium methyl complexes(LMgIand LMgMe,L=ligand 1),ligand 1 in the crude reaction mixture.This is different from the previously reportedβ-diketiminate ligandswhich upon reaction with one equiv.MeMgI produced the corresponding magnesium iodide complexes in good yield[8,20].Due to the fact that the crudemixturewasvery complex,the product couldnt be separated completely. We postulated that the resultant magnesium iodide complex(LMgI)is too reactive and further reacted with MeMgI immediately to generate the corresponding magnesium methyl complex(LMgMe).Hencewe added two equiv.MeMgI to the solution of ligand 1 in THF, the desired magnesium methyl complex 4 was indeed obtained as colorless crystals in good yield(72%) (Scheme 2).This offered an alternative method to synthesize the magnesium methyl complex which previously prepared from dimethylmagnesium with the relevantβ-diketiminate ligand[20-24].
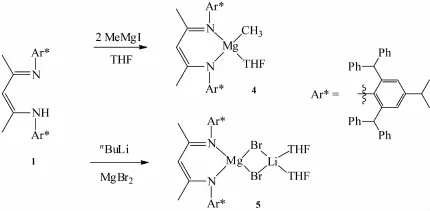
Scheme 2 Syntheses ofmagnesium complexes
NMR spectra of complex 4 displayed a very characteristic upfield Mg-CH3resonance atδ-0.81 (singlet,3H)in1H NMR spectrum and atδ-15.1 in the13C NMR spectrum respectively.These chemical shifts are similar to those observed for the related unsolvated dimeric Dipp-substitutedβ-diketiminate magnesium methyl complex[(DippNacnac)Mg(Me)]2(δ-1.17(1H),δ -18.6(13C))[19]and thoseofmonomeric three-coordinated tert-butyl-substitutedβ-diketiminato complex[(tBu Nacnac)Mg(Me)](δ-1.37(1H),δ-16.8(13C))[22]and 2,6-bis(diphenylmethyl)-p-tolyl-substitutedβ-diketiminate complex[(ArNacnac)Mg(Me)](ArNacnac=CH(C(Me)N (2,6-CHPh2-4-MeC6H2))2,δ-1.27(1H),δ-18.1(13C))[23].
Single crystals of complex 4 suitable for X-ray crystallographic analysis were obtained from THF/ hexanemixture at room temperature(Fig.3).Selected bond lengths and angles are listed in Table 3.The geometry at magnesium is distorted tetrahedral with angles in the range of 94.52(6)°~126.68(8)°.The Mg metal center is coordinated by two nitrogen atoms of ligand(N(1)and N(2)),one oxygen atom of THF(O(1)) and one carbon(C(76)).The Mg(1)-C(76)bond length (0.210 96(19)nm)in 4 falls in the range of 0.210 7(6)~0.218 9(4)nm in those similar solvatedβ-diketiminate magnesium methyl complexes.The bond angle of N(1)-Mg(1)-N(2)in 4 was 94.52(6)°,which is bigger than those similar solvatedβ-diketiminate magnesium methyl complexes reported previously(91.8(2)°~94.51(12)°)[21-23].
In order to get the desired magnesium halide complex,the ligand 1 was treated withnBuLi to produce the corresponding lithium salt at room temperature.Upon further reaction with magnesium bromide(MgBr2)in THF,the Mg-Libimetallic complex
5 wasobtained as colorless crystals in good yield(56%) (Scheme 2).This is quite different from the very similar 2,6-bis(diphenylmethyl)-p-tolyl-substitutedβ-diketiminate ligand which gave the corresponding magnesium bromide complex[(ArNacnac)MgBr(OEt2)]in very low yield(<5%)when treated with methylmagnesium bromide(MeMgBr)[23].However,the similar reaction between the potassium salt of ligand 1 andmagnesium chloride(MgCl2)in THF did not give the corresponding magnesium chloride complex since only the startingmaterial ligand 1 was recovered after workup.
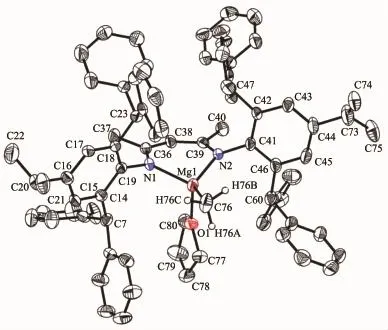
Fig.3 Molecular structure of 4 with thermal ellipsoids at the 50%probability level
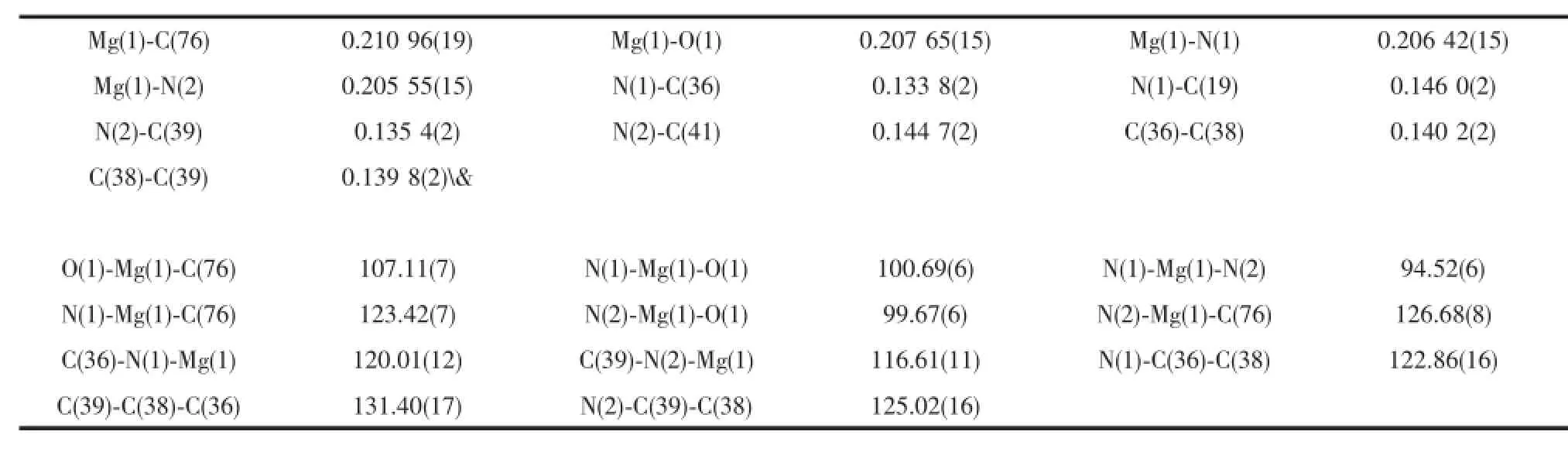
Table 4 Selected bond lengths(nm)and angles(ο)for complex 4
Single crystals of complex 5 suitable for X-ray crystallographic analysis were obtained from toluene/ hexanemixture at room temperature(Fig.4).Selected bond lengths and angles are listed in Table 4.Single crystal X-ray diffraction analysis of 5 showed that it was the Mg-Li bimetallic complex which is different from the normal monometallicβ-diketiminate magnesium halide complexes.The Mg,Li,Br(1)and Br(2)are almost in the rhombus plane.The geometry at magnesium is distorted tetrahedral with angles in the range of 96.62(15)°~118.89(11)°.The Mgmetal center is coordinated by two nitrogen atoms of ligand (N(1)and N(2))and two bromine atoms(Br(1)and Br (2)).The four-coordinated lithium is also distorted tetrahedral which is bound by two oxygen atoms of THF(O(1)and O(2))and two bromine atoms(Br(1) and Br(2)).The bond length of Mg-Br(0.250 67(15) and 0.250 45(15)nm)in 5 is slightly longer than that of very similar methyl-substitutedβ-diketiminate magnesium bromide complex[(ArNacnac)MgBr(OEt2)] (0.249 44(10)nm).The bond angle of N(1)-Mg(1)-N(2)
(96.62(15)°)in 5 is slightly smaller than thatofmethylsubstituted[(ArNacnac)MgBr(OEt2)](96.86(11)°)[23].
Finally,the correspondingmagnesium methyl and bromide complexes(4 and 5)as catalyst have been used in the hydrosilylation of acetophenone[25-27].At room temperature,using(EtO)3SiH as silane source in the presence of 10%(n/n)catalyst,the hydrosilylation reaction of acetophenone proceeded very slowly. However when the reaction temperature was increased to 80℃the reaction was completed in 24 h with 70% and 83%conversion for 4 and 5 respectively which was monitored by1H NMR spectroscopy in C6D6(Scheme 3).
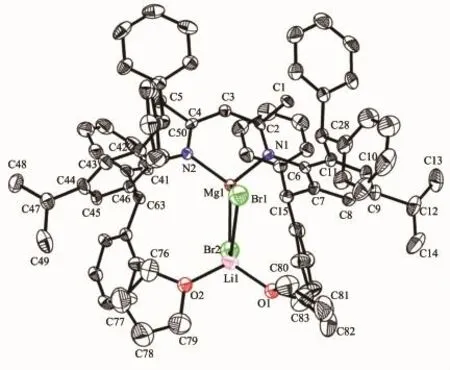
Fig.4 Molecular structure of 5 with thermal ellipsoids at the 50%probability level
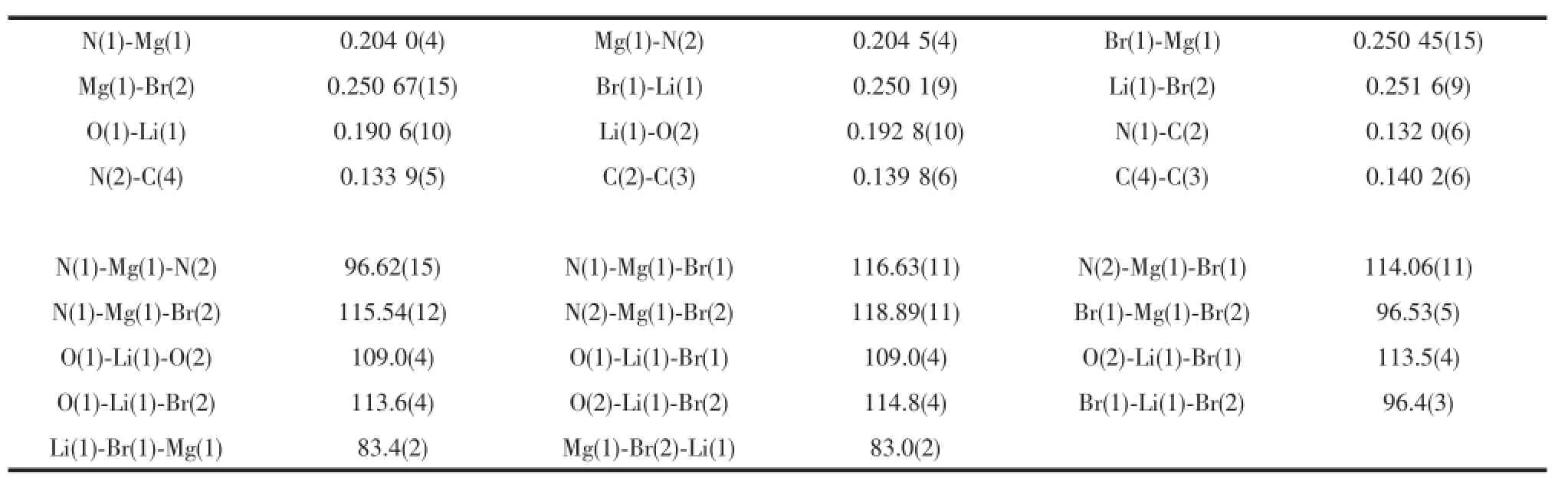
Table 5 Selected bond lengths(nm)and angles(°)for com p lex 5
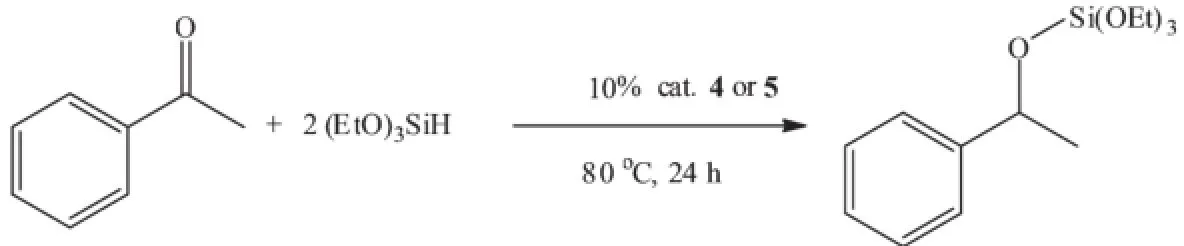
Scheme 3 Hydrosilylation of acetophenone by magnesium catalysts 4 and 5
3 Conclusions
In summary,we have successfully synthesized two new sterically bulkyβ-diketiminate ligands.They have been used to synthesize the corresponding
magnesium methyl and Mg-Li bimetallic magnesium bromide complexes which showed moderate catalytic activity in the hydrosilylation of acetophenone.Their molecular structures were confirmed by X-ray single crystal diffraction determination.The applications in the preparation of subvalent metal complexes and other applications are in progress in our group.
Acknow ledgem ents:Financial supports from the National Natural Science Foundation of China(Grants No.21372117, 21501094),the Natural Science Foundation of Jiangsu Province, China(Grants No.BK20141468,BK20130952,BK20140968), and the Qing Lan Project of Jiangsu Province are gratefully acknowledged.
[1]Bourget-Merle L,Lappert M F,Severn J R.Chem.Rev., 2002,102:3031-3065
[2]Sarish S P,Nembenna S,Nagendran S,et al.Acc.Chem. Res.,2011,44:157-170
[3]Holland PL.Acc.Chem.Res.,2015,48:1696-1702
[4]Parks JE,Holm R H.Inorg.Chem.,1968,7:1408-1416
[5]Liu P,Chen H,Zhang Y,et al.Dalton Trans.,2014,43: 5586-5594
[6]Wang Y,Quillian B,Wei P,et al.J.Am.Chem.Soc.,2005, 127:11944-11945
[7]Green S P,Jones C,Stasch A.Science,2007,318:1754-1757
[8]Bonyhady S J,Jones C,Nembenna S,et al.Chem.Eur.J., 2010,16:938-955
[9]Stasch A,Jones C.Dalton Trans.,2011,40:5659-5672
[10]Jones C,Stasch A.Top.Organomet.Chem.,2013,45:73-102
[11]Ma M,Wang W,Yao W.Chin.J.Org.Chem.,2016,36:72-82
[12]Asay M,Inoue S,Driess M.Angew.Chem.Int.Ed.,2011, 50:9589-9592
[13]Rekken BD,Brown TM,Fettinger JC,et al.J.Am.Chem. Soc.,2012,134:6504-6507
[14]Had lington T J,Li J,Jones C.Can.J.Chem.,2014,92:427-433
[15]Dierick S,Dewez D F,Marko I.E.Organometallics,2014, 33:677-683
[16]Sheldrick GM.SADABS,University of Göttingen,Germany, 1997.
[17]Sheldrick G M.SHELXL-97,University of Göttingen, Germany,1997.
[18]WeberskiM P,Mclauchlan C C.J.Coord.Chem.,2008,61: 2371-2379
[19]Bailey P J,Lidd le ST,Parsons S.Acta Crystallogr.Sect.E, 2001,57:o661-o662
[20]Dove A P,Gibson V C,Hormnirun P,et al.Dalton Trans., 2003,15:3088-3097
[21]Gibson V C,Segal JA,White A JP,et al.J.Am.Chem. Soc.,2000,122:7120-7121
[22]Bailey P J,Coxall R A,Dick C M,et al.Organometallics, 2001,20:798-801
[23]Arrowsmith M,Maitland B,Kociok-Köhn G,et al.Inorg. Chem.,2014,53:10543-10552
[24]Bailey P J,Dick C M E,Fabre S,et al.Dalton Trans., 2000,10:1655-1661
[25]Bleith T,Wadepohl H,Gade LH.J.Am.Chem.Soc.,2015, 137:2456-2459
[26]Hashimoto T,Urban S,Hoshino R,et al.Organometallics, 2012,31:4474-4479
[27]Yang J,Tilley TD.Angew.Chem.Int.Ed.,2010,49:10186-10188
Sterically Bulkyβ-Diketim inate Magnesium Com p lexes:Syntheses,Crystal Structure and Catalytic Hydrosilylation
MA Meng-Tao*,1SHEN Xing-Chao1YU Zhi-Juan1YAOWei-Wei2DU Li-Ting1XU Li*,1
(1College of Science,Nanjing Forestry University,Nanjing 210037,China)
(2College of Pharmacy,Nanjing University of Chinese Medicine,Nanjing 210023,China)
Two new sterically bulkyβ-diketiminate compounds CH(C(Me)N(2,6-CHPh2-4-iPrC6H2))2H(1)and(CH (C(Me)N(2,4-C(CH3)3C6H2))2H(2)were synthesized by the reaction of 2,4-pentanedione with the corresponding primary amines.Treatment of ligand 1 with Grignard reagent MeMgI in THF provided a four-coordinated magnesium methyl complex.The lithium salt of 1 was subsequently reacted with MgBr2in THF to yield the Mg-Li bimetallic magnesium bromide complex.The molecular structures of twoβ-diketiminate ligands and their corresponding magnesium methyl and bromide complexes have been characterized using X-ray crystallography. The corresponding magnesium methyl and bromide complexes have shown moderate catalytic activity in the hydrosilylation of acetophenone.CCDC:1445844,1;1445845,2;1480136,4;1480137,5.
magnesium;β-diketim inate ligand;crystal structure;hydrosilylation
2;O614.111
A
1001-4861(2016)10-1857-10
10.11862/CJIC.2016.232
2016-08-04。收修改稿日期:2016-09-12。
国家自然科学基金(No.21372117,21501094)和江苏省自然科学基金(No.BK20141468,BK20130952,BK20140968)资助项目。
*通信联系人。E-mail:mengtao@njfu.edu.cn,xuliqby@njfu.edu.cn;会员登记号:S06N1815M1608(马猛涛)。
猜你喜欢
南京林业大学学报(自然科学版)(2023年1期)2023-03-30 03:40:00
南京林业大学学报(自然科学版)(2022年3期)2022-11-30 01:46:54
南京林业大学学报(自然科学版)(2022年5期)2022-10-19 02:07:38
高等理科教育(2022年2期)2022-05-09 07:34:50
南京林业大学学报(自然科学版)(2021年5期)2021-10-10 06:27:44
橡胶工业(2015年2期)2015-07-29 08:29:46
西南军医(2014年5期)2014-04-25 07:42:49
食品工业科技(2014年9期)2014-03-11 18:15:39
当代化工研究(2013年6期)2013-08-15 00:56:11
天然产物研究与开发(2012年4期)2012-12-22 09:01:16