
高效可见光响应Zn0.11Sn0.12Cd0.84S1.12/g-C3N4异质结光催化剂的制备及性能
2016-12-02 03:03胡绍争李法云范志平刘道胜
高等学校化学学报 2016年3期
张 倩, 胡绍争, 李法云, 范志平, 王 琼, 王 菲, 李 薇, 刘道胜
(辽宁石油化工大学化学化工与环境学部, 抚顺113001)
高效可见光响应Zn0.11Sn0.12Cd0.84S1.12/g-C3N4异质结光催化剂的制备及性能
张 倩, 胡绍争, 李法云, 范志平, 王 琼, 王 菲, 李 薇, 刘道胜
(辽宁石油化工大学化学化工与环境学部, 抚顺113001)
以双氰胺、 醋酸锌、 四氯化锡、 醋酸镉和硫化钠为原料, 采用水热法制备了三元金属复合硫化物Zn0.11Sn0.12Cd0.84S1.12(ZnSnCdS)及一系列异质结催化剂ZnSnCdS/g-C3N4. 采用X射线衍射仪(XRD)、 扫描电子显微镜(SEM)、 紫外-可见光谱仪(UV-Vis)、 傅里叶变换红外光谱仪(FTIR)、 电感耦合等离子体-质谱仪(ICP-MS)、 荧光光谱仪(PL)和X射线光电子能谱仪(XPS)等对催化剂进行了表征. 结果表明, ZnSnCdS与g-C3N4之间以C—S键紧密结合, 构筑了异质结, 促进了界面电荷迁移, 抑制了光生电子-空穴对的复合. 可见光下降解染料罗丹明B(RhB)的结果表明, ZnSnCdS/g-C3N4异质结催化剂的光催化性能与单纯g-C3N4, ZnSnCdS及双组分硫化物/g-C3N4异质结催化剂相比均有大幅度提高, ZnSnCdS与g-C3N4质量比为4∶1时异质结催化剂表现出最大的速率常数(0.1508 min-1), 是单纯g-C3N4和ZnSnCdS的32.3倍和4.9倍. 其它三元金属复合硫化物如ZnMoCdS, MoNiCdS和NiSnCdS与g-C3N4之间也能有效形成异质结, 促进电子-空穴对的分离和催化性能的提升.
石墨型氮化碳; 三元金属硫化物; 异质结催化剂; 光催化; 可见光
目前, 环境污染和能源危机日益严重, 发展可再生能源受到广泛重视. 太阳能是可再生能源, 光催化技术利用太阳能来驱动一系列化学反应, 如将太阳能转化为化学能或降解有机污染物, 在解决环境污染和能源危机方面表现出巨大的潜力. 但是, 常见的光催化剂对太阳光中可见光的利用率较低, 在很大程度上限制了其实际应用[1,2]. 近年来, 石墨型氮化碳(g-C3N4)由于具有优异的化学稳定性和独特的电子能带结构, 作为可见光催化剂在制氢[3]、 有机合成[4]、 分解污染物[5]等领域显现出优越的催化性能. g-C3N4的带隙能较窄(2.7 eV), 可吸收波长450 nm以下的可见光. 然而, g-C3N4光生载流子寿命短, 易复合, 导致量子效率较低[6].
开发g-C3N4基异质结光催化剂, 使光生电荷通过异质结快速有效地转移是解决上述问题的有效途径之一. 设计合适的异质结对提高光催化性能有显著影响[7]. 纳米结构的金属硫化物在光催化领域备受关注, 主要有单组分硫化物ZnS, CdS, In2S3, SnS2, PbS[8~12]等和双组分硫化物ZnxCd1-xS[13], ZnIn2S4[11], CuInS2[14]等. 这些金属硫化物一般具有较窄的禁带宽度, 能够吸收可见光. 另一方面, 多种金属硫化物的价带与导带的能级位置与g-C3N4的能级位置相互匹配[15], 能满足形成异质结催化剂的基本条件. 因此, 许多研究者采用金属硫化物来提高g-C3N4的光生电荷-空穴分离效率. Ge等[16]采用浸渍-沉积法合成了g-C3N4/MoS2复合催化剂, 发现层状MoS2均匀地附着在g-C3N4表面并形成异质界面, 有效促进了光生电荷转移; 同时g-C3N4/MoS2复合催化剂在可见光照射下能够增强析氢活性, g-C3N4/MoS2质量比为199∶1时效果最好, 析氢速率达到23.10 μmol/h, 是纯g-C3N4的11.3倍. Sun等[17]合成了g-C3N4/Zn0.25Cd0.75S复合催化剂, 并用于光催化还原水中的Cr(Ⅵ)离子和降解罗丹明B(RhB). 结果表明, 异质结的形成加速了光生电子-空穴对的分离, RhB的光降解效率也相应地提高, g-C3N4/Zn0.25Cd0.75S 质量比为1∶4(CNZS-20)时, 复合催化剂45 min对RhB的降解率高达99%. 研究发现, 在CNZS-20复合催化剂表面, g-C3N4和Zn0.25Cd0.75S的晶面紧密接触并构筑成异质结界面, 可为电荷转移提供良好的条件. 同时, CNZS-20复合催化剂在25 min内对水中Cr(Ⅵ)离子的还原率达到99%, 这主要是由于异质结的形成促进了电荷转移, 使Cr(Ⅵ)被还原为Cr(Ⅲ)离子.
本文以双氰胺、 醋酸锌、 四氯化锡、 醋酸镉和硫化钠为原料, 采用水热法制备了一系列ZnSnCdS/g-C3N4异质结催化剂, 以RhB的降解为探针反应考察了催化剂在可见光下的光催化性能, 并探讨了异质结对催化剂的结构、 光学性质及光催化性能的影响.
1 实验部分
1.1 试剂与仪器
双氰胺和醋酸锌(天津化学试剂厂), 四氯化锡(天津市永大化学试剂有限公司); 醋酸镉(天津市光复精细化工研究所); 硫化钠(天津市恒兴化学试剂制造有限公司). 以上试剂均为分析纯, 使用前未经提纯. 所有水溶液均用去离子水配制.
日本岛津公司XRD-7000 X射线衍射仪(CuKα1辐射源, 工作电压40 kV, 工作电流30 mA); Micrometrics ASAP 2010物理吸附仪(在-196 ℃下进行测试, 测试前样品先于350 ℃下真空脱气处理10 h), 样品的比表面积通过Brunauer-Emmett-Teller(BET)方法求得; 美国Nicolet公司Nicolet360傅里叶变换红外光谱仪(FTIR); 日本电子有限公司JSM 5600LV扫描电子显微镜(SEM). 美国Perkin Elmer公司Elan6100DRC型电感耦合等离子体质谱仪(ICP-MS). 日本JASCA公司UV-550紫外-可见光谱仪; 赛默飞世尔科技有限公司Thermo ESCALAB 250 X射线光电子能谱仪(XPS); 日本分光公司FP-6300荧光分光光度计(以Xe灯为激发光源).
1.2 ZnSnCdS/g-C3N4复合催化剂的制备
根据文献[18]中的方法, 采用双氰胺为前驱体制备g-C3N4, 记为CN. 称取一定量醋酸锌、 四氯化锡与醋酸镉(Zn/Sn/Cd摩尔比1∶1∶8)溶于10 mL去离子水中, 搅拌15 min后, 将一定量的CN(0.1, 0.2, 0.5, 0.8 g)加入上述溶液中, 超声分散30 min, 在搅拌条件下缓慢滴加20 mL 0.5 mol/L的硫化钠溶液. 得到的悬浊液搅拌12 h后, 装入反应釜中于160 ℃反应16 h. 待反应釜冷却至室温后将产物离心分离, 用去离子水和无水乙醇洗涤数次, 再于80 ℃下干燥12 h, 即得到一系列ZnSnCdS/g-C3N4异质结催化剂, 记为ZnSnCdS-CN(x), 其中x为CN的质量分数. 重复上述制备过程, 在不加g-C3N4条件下, 得到的催化剂命名为ZnSnCdS. 上述合成方法及金属摩尔比不变, 将3种金属盐两两组合, 制备的催化剂命名为ZnSnS-CN(20%), ZnCdS-CN(20%)和SnCdS-CN(20%). 按照文献[17,19,20]方法合成催化剂Zn0.25Cd0.75S-CN(20%), Zn0.8Cd0.2S-CN(50%)和Zn0.28Cd0.72S-CN(70%).
1.3 光催化活性评价
以250 W高压钠灯(主波长400~800 nm)为可见光光源, 以0.5 mol/L的亚硝酸钠水溶液为钠灯冷却循环水滤去光源中的紫外光, 以染料RhB为降解物评价催化剂的光催化活性. 将0.1 g催化剂加入到500 mL 10 mg/L的RhB溶液中, 搅拌0.5 h达到催化剂对反应底物的吸附平衡. 搅拌下将溶液放在光源下进行照射, 同时鼓入空气(30 ℃, 标准大气压). 每隔0.5 h取5 mL悬浊液置于离心管中, 在3000 r/min的转速下离心5 min后取上层清液, 用紫外-可见分光光度计测定550 nm处的吸光度.
2 结果与讨论
2.1 催化剂的表征

Fig.1 XRD patterns of CN, ZnSnCdS and ZnSnCdS-CN(x)a. CN; b. ZnSnCdS; c. ZnSnCdS-CN(10%); d. ZnSnCdS-CN(20%); e. ZnSnCdS-CN(50%).
2.1.1 XRD表征 图1为CN, ZnSnCdS和ZnSnCdS-CN(x)的XRD谱图. 由图1可见, g-C3N4有2个特征峰, 分别位于2θ=13.1°和27.5°处, 其中27.5°处的衍射峰较强, 为芳香族化合物层间堆积特征峰, 晶面指数标记为(002), 对应层间距d=0.33 nm, 说明g-C3N4具有与石墨类似的层状结构; 13.1°处的衍射峰是芳环体系的层间堆垛结构(melon类结构)的特征峰, 晶面指数标记为(100), 对应3-s-三嗪结构中氮孔结构间距d=0.67 nm[21]. ZnSnCdS有3个特征峰, 分别位于2θ=26.9°, 44.4°和52.4°处, 与立方相CdS的特征峰位置(26.6°, 43.9°和52.0°)很相近[22]. 此外, 谱图中未发现SnS2和ZnS的特征衍射峰[23,24], 这可能是由于Zn和Sn掺入CdS的晶格得到了三元金属复合硫化物Zn0.1Sn0.1Cd0.8S1.1, 而非单纯ZnS, SnS2和CdS的混合物. Zn和Sn的掺杂作用使CdS发生晶格扭曲, 衍射峰位置略有偏移. 与ZnSnCdS相比, ZnSnCdS/g-C3N4异质结催化剂的衍射峰位置没有发生变化, 这是由于CdS在26.6°处的衍射峰与g-C3N4在27.5°处的衍射峰相互叠加所致. Ge等[25]也报道了相似的结果.
2.1.2 SEM表征 如图2(A)所示, g-C3N4具有无规则的类似于石墨的光滑层状结构. 由图2(B)和(C)中可以清楚地看出, ZnSnCdS具有类似花状结构, 每个花瓣都是形状规整且厚度均一的纳米片. 由图2(D)可见, 在ZnSnCdS-CN(20%)中, ZnSnCdS破碎成片状结构, 并附着在g-C3N4的表面. 这种2D/2D的面-面接触方式有利于形成异质结, 可促进界面电荷迁移, 抑制光生电子-空穴对的复合. ICP结果显示, ZnSnCdS-CN(20%)中Zn, Sn, Cd, S的质量分数分别为3.6%, 7.2%, 51.2%和18.0%, 与理论值非常接近. 根据ICP结果计算得出, 三元金属硫化物的实际组成为Zn0.11Sn0.12Cd0.84S1.12.

Fig.2 SEM images of synthesized CN(A), ZnSnCdS(B, C) and ZnSnCdS-CN(20%)(D)
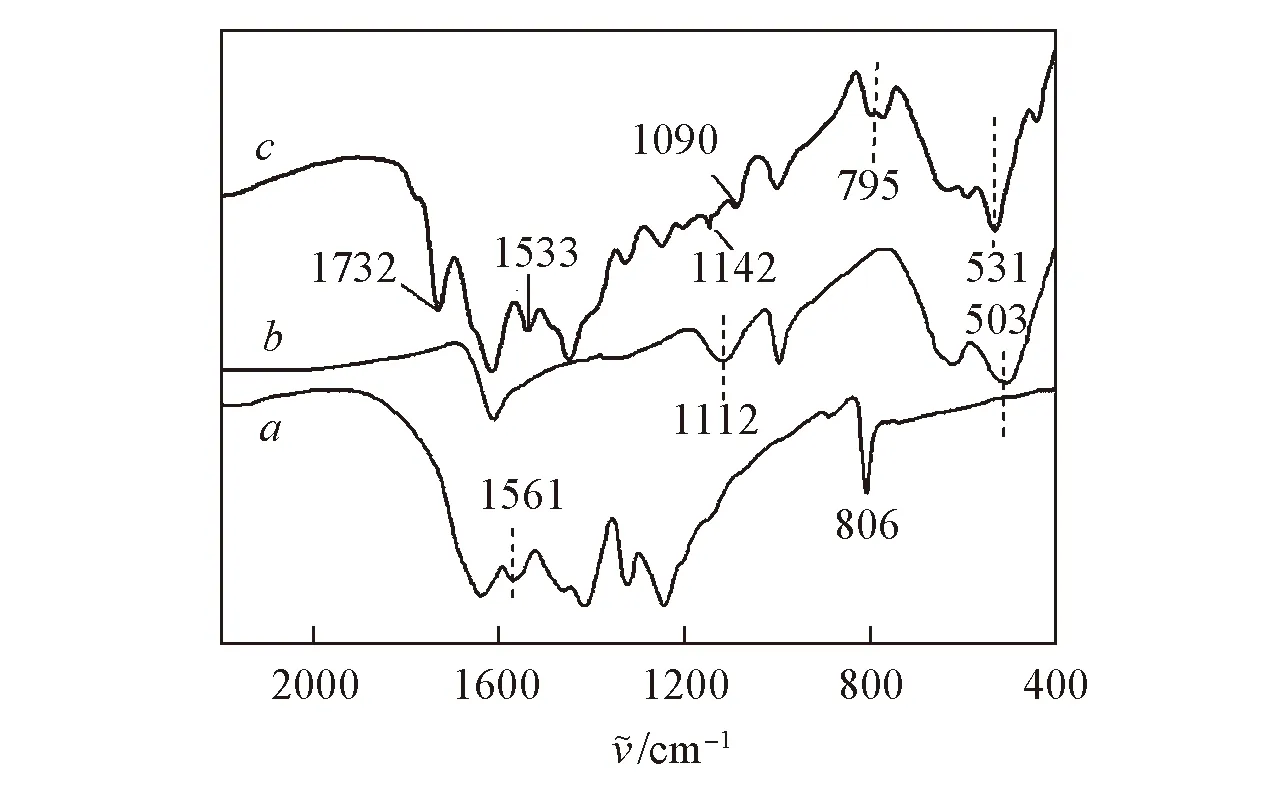
Fig.3 FTIR spectra of CN(a), ZnSnCdS(b) and ZnSnCdS-CN(20%)(c)

2.1.4 UV-Vis光谱表征 UV-Vis光谱图能直观地反映所制备催化材料的光吸收特性. 如图4所示, g-C3N4的吸收边约为460 nm. 根据Kubelka-Munk函数法[25]计算出其带隙能约为2.69 eV; ZnSnCdS的吸收边界为614 nm, 对应的带隙能为2.02 eV. 根据文献[15,22,26,30]报道, ZnS, SnS2和CdS的带隙能分别为3.4, 2.2和2.4 eV, 均大于制备的三元金属硫化物ZnSnCdS. 此结果进一步证实了制备的催化剂不是单纯的ZnS, SnS2和CdS的混合物, 而是三元金属复合硫化物Zn0.11Sn0.12Cd0.84S1.12. 金属的掺杂作用使得原有的金属硫化物的电子结构发生变化, 导致其光学性质也发生变化. 制备的系列ZnSnCdS/g-C3N4异质结催化剂的吸收边界位于g-C3N4和ZnSnCdS之间, 证明两组分间存在电子耦合作用, 并由此形成异质结. Liu等[31]和Dong等[32]也得到了类似的实验结果. 计算得到ZnSnCdS-CN(10%), ZnSnCdS-CN(20%), ZnSnCdS-CN(50%)和ZnSnCdS-CN(80%)的带隙能分别为2.08, 2.13, 2.23和2.47 eV. 此外, ZnSnCdS/g-C3N4异质结催化剂在全波长范围内的吸收强度均高于g-C3N4. 这说明制备的ZnSnCdS/g-C3N4异质结催化剂可吸收比单纯g-C3N4催化剂更多的光能, 产生更多的光生电子-空穴对, 因此更有利于光催化反应的发生.
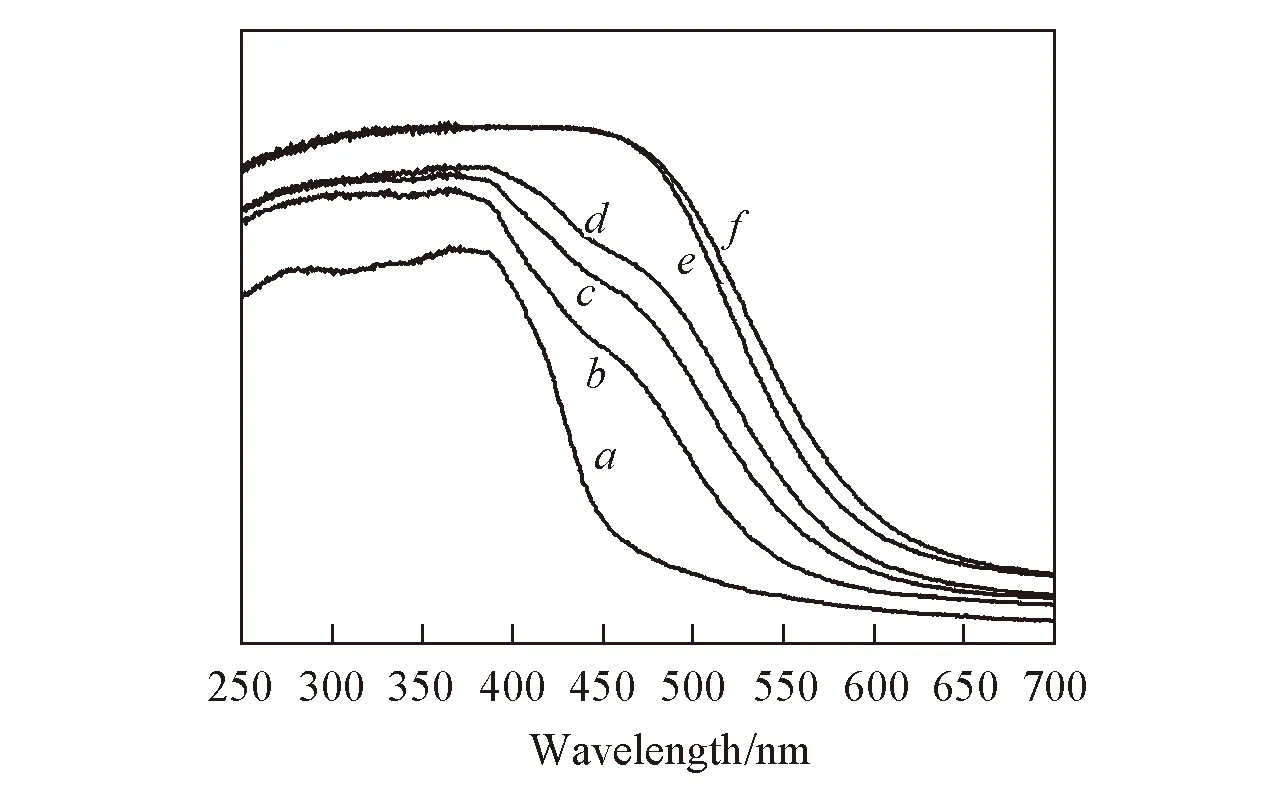
Fig.4 UV-Vis spectra of CN, ZnSnCdS and ZnSnCdS-CN(x)a. CN; b. ZnSnCdS-CN(80%); c. ZnSnCdS-CN(50%); d. ZnSnCdS-CN(20%); e. ZnSnCdS-CN(10%); f. ZnSnCdS.

Fig.5 Schematic illustration of electron-hole separation and transport at the g-C3N4/ZnSnCdS heterojunction interface
根据文献[33]报道, 半导体材料的价带能级(EVB)可以利用公式EVB=X-E-1/2Eg[其中X为半导体的绝对电负性,E为氢标下自由电子对的能量(约为4.5 eV),Eg为半导体带隙能]计算得到. 计算结果显示, Zn0.1Sn0.1Cd0.8S1.1的EVB为1.86 eV. 相应的导带能级ECB为-0.37 eV. g-C3N4的ECB和EVB分别为-1.12和1.57 eV[34]. 可见, ZnSnCdS和g-C3N4的能级匹配较好. 如图5所示, 当二者组成异质结催化剂后, 在电势差驱动力的作用下, 光电子能快速地从g-C3N4转移至ZnSnCdS表面, 而空穴会从ZnSnCdS向g-C3N4聚集. 电子和空穴重新分布后, 会在两组分间的异质界面建立一个稳定的内电场, 抑制电子-空穴的复合过程, 使量子效率和光催化性能得到极大提升.
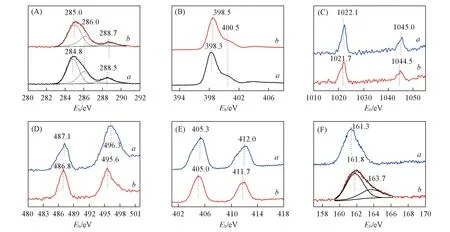
Fig.6 XPS spectra of as-prepared catalysts in the region of C1s(A), N1s(B), Zn2p(C), Sn2p(D), Cd2p(E) and S2p(F)(A), (B) a. CN; b. ZnSnCdS-CN(20%). (C)—(F) a. ZnSnCdS; b. ZnSnCdS(20%)-CN.
图6(C)~(E)分别为所制备催化剂的Zn2p, Sn2p和Cd3d轨道的XPS谱图. 可见, ZnSnCdS中Zn2p轨道的结合能位于1022.1和1045.0 eV处, Sn2p轨道的结合能位于487.1和496.3 eV处, Cd3d轨道的结合能位于405.3和412.0 eV处, 明显区别于单纯ZnS, SnS2和CdS在相应轨道处的结合能[29,36~38]. 这也再次证实制备的催化剂不是单纯ZnS, SnS2和CdS的混合物, 而是三元金属复合硫化物Zn0.11Sn0.12Cd0.84S1.12, 金属间的相互作用导致了结合能的差异. 对于ZnSnCdS-CN(20%), 其Zn2p, Sn2p和Cd3d轨道的结合能与ZnSnCdS相比均向低结合能方向偏移. 这一结果再次证实了g-C3N4与ZnSnCdS之间存在较强的相互作用. 如图6(F)所示, ZnSnCdS样品中S2p轨道的结合能位于161.3 eV处, 说明存在金属—S键, 硫的价态为S2-. ZnSnCdS-CN(20%)样品中, 除了金属—S键, 硫还表现出另外一种存在形式, 其结合能位于163.7 eV处, 应归属于g-C3N4与ZnSnCdS之间形成的C—S键[39]. 此结果与FTIR结果相一致.

Fig.7 Photoluminescence(PL) emission spectra of as-prepared catalystsa. CN; b. ZnSnCdS; c. ZnSnCdS-CN(80%); d. ZnSnCdS-CN(50%); e. ZnSnCdS-CN(10%); f. ZnSnCdS-CN(20%).
2.1.6 PL光谱表征 光生电子-空穴对的分离效率对催化剂的性能十分重要, 光生载流子的分离可以保证光催化反应有效进行[40]. 图7为所制备催化剂的PL谱图, 激发光源波长为320 nm. 可见, g-C3N4在460 nm附近有很强的荧光发射峰, 与UV-Vis谱图(图4)中g-C3N4的吸收边一致. ZnSnCdS-CN(x)系列样品的荧光光谱发生明显的荧光猝灭现象, 说明g-C3N4和ZnSnCdS两组分形成异质结后能有效抑制电子-空穴对的复合. PL光谱的峰强度顺序为CN>ZnSnCdS>ZnSnCdS-CN(80%)>ZnSnCdS-CN(50%)>ZnSnCdS-CN(10%)>ZnSnCdS-CN(20%), 可见, ZnSnCdS-CN(20%)表现出最佳的电子-空穴对的分离效率.
2.2 光催化活性评价
CN, ZnSnCdS和ZnSnCdS-CN(x)在可见光下对RhB的降解反应结果如图8(A)所示. 由图8(A)可以看出, CN在120 min内对RhB的降解率为43%, ZnSnCdS在120 min内对RhB的降解率为97.9%. ZnSnCdS-CN(x)系列催化剂的光催化性能与g-C3N4和ZnSnCdS相比均有显著提高, 这是由于g-C3N4与ZnSnCdS复合后形成异质结界面, 促进了光生电子-空穴对的有效分离. 此外, ZnSnCdS-CN(x)系列催化剂对RhB的吸附能力与2个单组分相比也有显著提高. 由图8(A)可见, ZnSnCdS-CN(20%)展示了最佳的光催化性能, 在30 min内对RhB的降解率达到100%. 根据上述实验结果可知, ZnSnCdS/CN质量比为4∶1时催化性能最佳, 原因可能为: (1) 氮气吸附结果显示, ZnSnCdS和CN的比表面积分别为58和9.5 m2/g. ZnSnCdS-CN(20%)样品中ZnSnCdS较多可以增加催化剂对反应底物的吸附能力; (2) UV-Vis光谱结果表明, ZnSnCdS对可见光的吸收能力远高于CN, 因此ZnSnCdS较多也有利于复合催化剂对可见光的利用. 然而对于ZnSnCdS-CN(10%), 由于质量比及比表面积的较大差距, 导致两组分间形成的异质结面积有所下降, 因此导致催化性能降低.
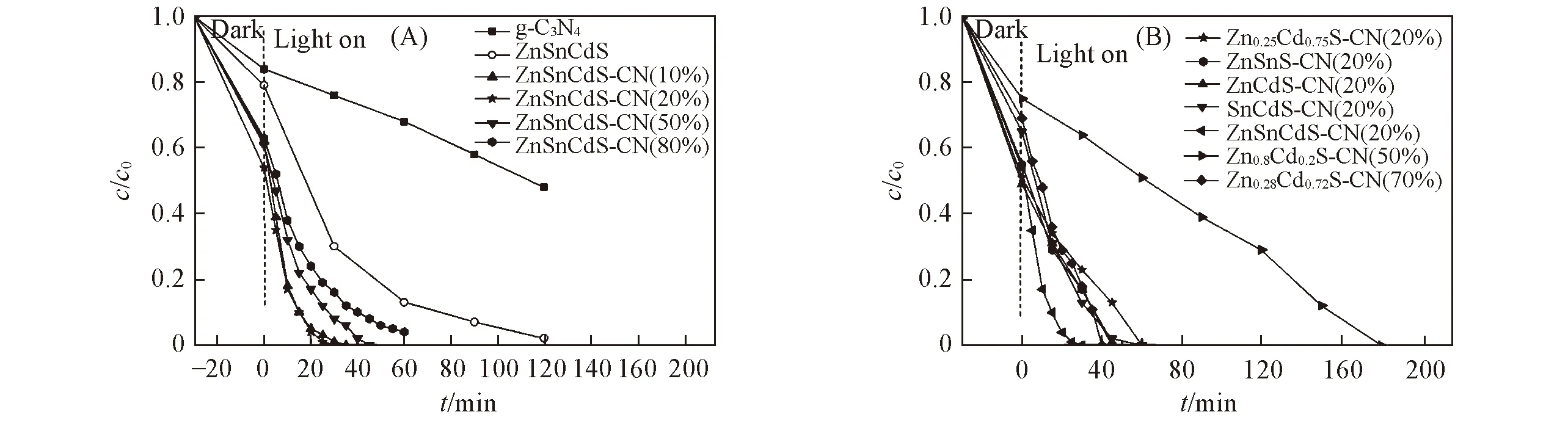
Fig.8 Photocatalytic RhB degradation performances of as-prepared catalysts under visible light irradation
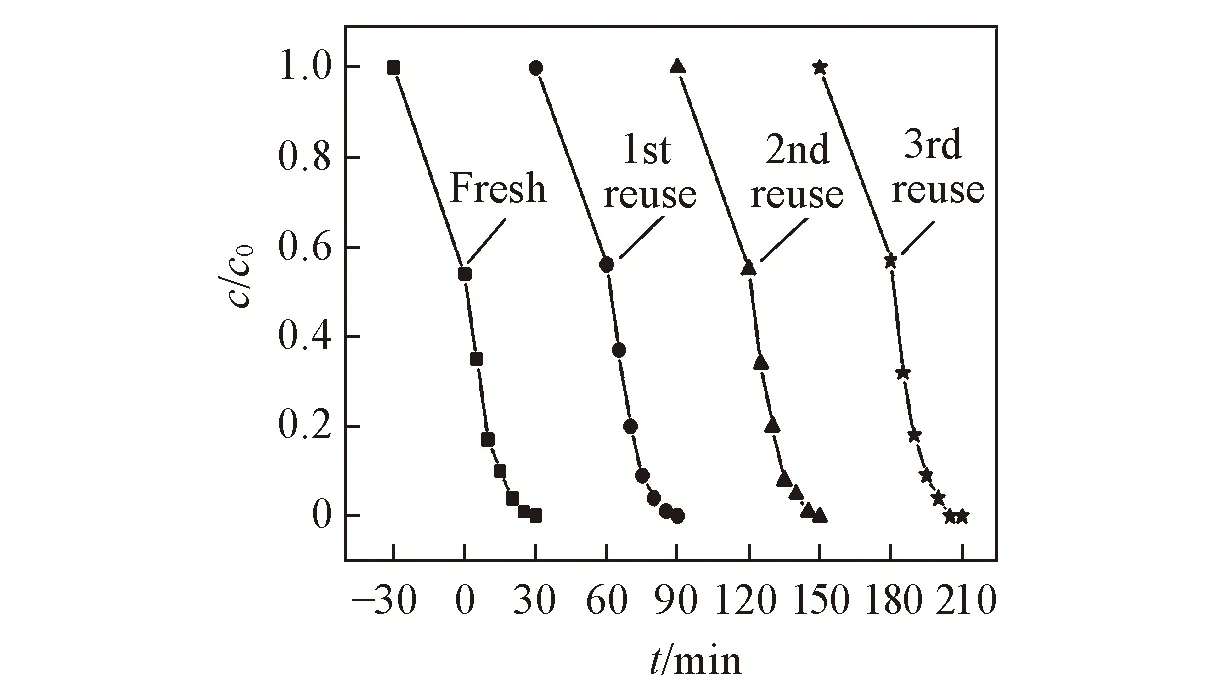
Fig.9 Photocatalytic stability of ZnSnCdS-CN(20%) for RhB degradation under visible light
通常, 光降解反应遵循一级反应动力学, 动力学方程为-ln(c/c0)=kt(其中c和c0分别代表任意时刻t和初始时刻RhB的浓度, 由直线的斜率可得到速率常数k[41,42]). 计算结果表明, g-C3N4和ZnSnCdS存在下RhB光降解反应的速率常数分别为0.0047和0.0307 min-1. 异质结催化剂ZnSnCdS-CN(10%), ZnSnCdS-CN(20%), ZnSnCdS-CN(50%)和ZnSnCdS-CN(80%)存在下RhB光降解反应的速率常数分别为0.1353, 0.1508, 0.0754和0.0462 min-1, 其中ZnSnCdS-CN(20%)存在下RhB光降解反应表现出最大的速率常数, 是g-C3N4和ZnSnCdS存在下的32.3倍和4.9倍. 图8(B)对比了ZnSnCdS-CN(20%), ZnSnS-CN(20%), ZnCdS-CN(20%), SnCdS-CN(20%)和按照文献[17,19,20]的方法制备的3个催化剂对RhB降解的光催化性能, 可以看出, ZnSnS-CN(20%), ZnCdS-CN(20%)和SnCdS-CN(20%)的活性差别不大, 30 min时RhB的降解率均在80%左右, 明显低于ZnSnCdS-CN(20%). 可见, 三组分金属硫化物与g-C3N4组成的异质结催化剂比双组分金属硫化物与g-C3N4组成的异质结催化剂催化性能有显著提升. ZnSnCdS-CN(20%)的性能也明显优于文献报道的3个催化剂.
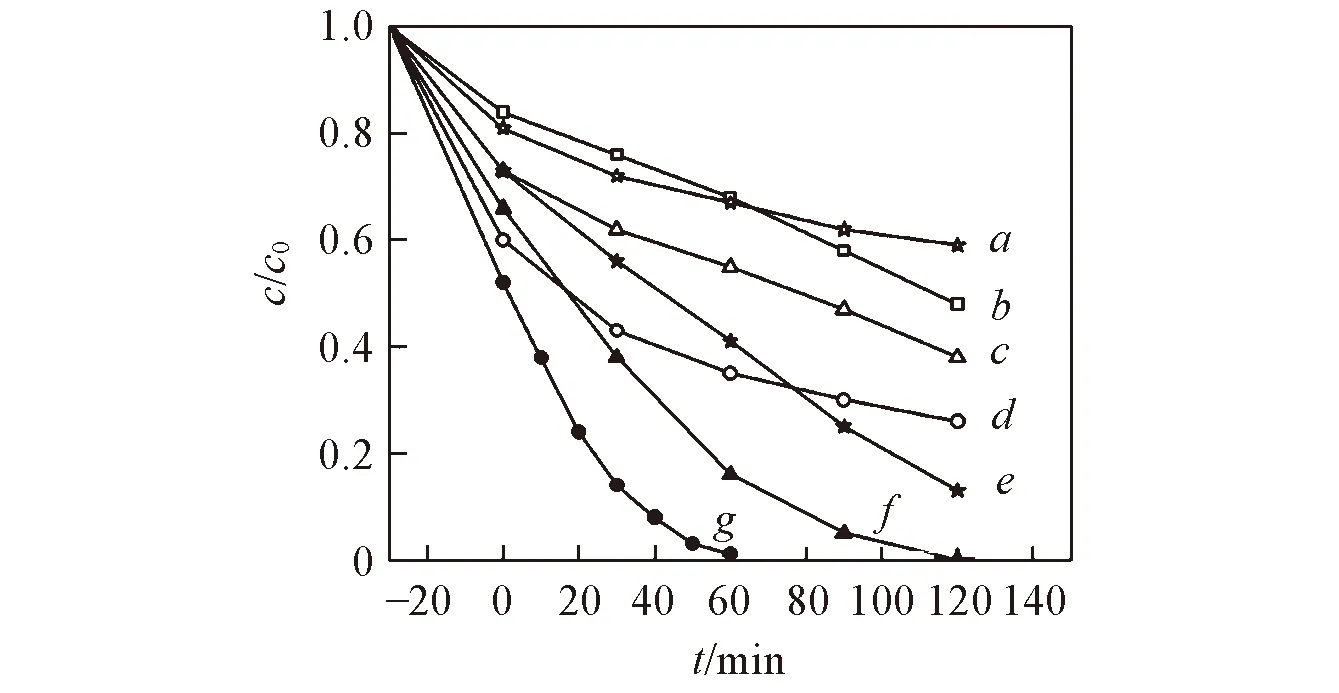
Fig.10 Photocatalytic RhB degradation performance over other three ternary metal sulfide/g-C3N4 catalystsa. NiSnCdS; b. g-C3N4; c. MoNiCdS; d. ZnMoCdS; e. NiSnCdS-CN(20%); f. MoNiCdS-CN(20%); g. ZnMoCdS-CN(20%).
ZnSnCdS-CN(20%)循环使用3次的结果如图9所示. 可以看出, 催化剂在3次循环使用后活性没有明显的降低, 说明ZnSnCdS-CN(20%)具有优越的催化稳定性. 此外, ICP 结果显示, 重复使用3次后ZnSnCdS-CN(20%)中Zn, Sn, Cd和S的质量分数分别为3.5%, 7.3%, 50.6%和18.4%, 与新制备的催化剂非常接近. 此外, 采用相似的水热法和金属摩尔比制备了其它几种三元金属复合硫化物ZnMoCdS, MoNiCdS和NiSnCdS及相应的异质结催化剂ZnMoCdS-CN(20%), MoNiCdS-CN(20%)和NiSnCdS-CN(20%). 可见光下对RhB的降解反应结果(图10)表明, ZnMoCdS-CN(20%)、 MoNiCdS-CN(20%)和NiSnCdS-CN(20%)的催化性能均优于相应的单组分催化剂, 其催化RhB降解反应的速率常数分别为0.0732, 0.0567和0.0143 min-1, 是相应单组分三元金属硫化物的14.0, 10.6和5.6倍, 是纯g-C3N4的15.7, 12.1和3.1倍. 此结果说明ZnSnCdS和其它三元金属复合硫化物与g-C3N4之间能有效构筑异质结, 促进电子-空穴对的分离.
综上所述, 以双氰胺、 醋酸锌、 四氯化锡、 醋酸镉和硫化钠为原料, 采用水热法制备了一系列ZnSnCdS/g-C3N4异质结催化剂. ZnSnCdS与g-C3N4之间以C—S键紧密结合, 形成异质结, 促进了界面电荷迁移, 抑制了光生电子-空穴对的复合, 使量子效率和光催化性能得到极大提升. 在可见光下对RhB的降解反应结果显示, ZnSnCdS-CN(x)系列催化剂的光催化性能与g-C3N4和ZnSnCdS相比均有显著提高, ZnSnCdS-CN(20%)表现出最佳的催化效果(速率常数=0.1508 min-1), 是g-C3N4和ZnSnCdS的32.3和4.9倍. 不仅ZnSnCdS, 其它三元金属复合硫化物, 如ZnMoCdS, MoNiCdS和NiSnCdS与g-C3N4之间也能有效构筑异质结, 促进电子-空穴对的分离和催化性能的提升.
[1] Kondo K., Murakami N., Ye C., Tsubota T., Ohno T.,Appl.Catal.B:Environ., 2013, 142/143(1), 362—367
[2] HARI Bala, Guo J. Y., Aruna., Yuan G. Y., Zhang Z. Y., Liu Z. R.,Chem.J.ChineseUniveisities, 2012, 33(12), 2716—2721(哈日巴拉, 郭金毓, 阿茹娜, 原光瑜, 张占营, 刘宗瑞. 高等学校化学学报, 2012, 33(12), 2716—2721)
[3] Zhang G. G., Zhang M. W., Ye X. X., Qiu X. Q., Lin S., Wang X. C.,Adv.Mater., 2014, 26(5), 805—809
[4] Xu J., Wu H. T., Wang X., Xue B., Li Y. X., Cao Y.,Phys.Chem.Chem.Phys., 2013, 15(13), 4510—4517
[5] Ge L.,MaterLett., 2011, 65(17/18), 2652—2654
[6] Niu P., Zhang L., Liu G., Cheng H.,Adv.Funct.Mater., 2012, 22(22), 4763—4770
[7] Zhang Q., Wang H. Y., Hu S. Z., Lu G., Bai J., Liu D., Gui J. G.,RSCAdv., 2015, 5(1), 42736—42743
[8] Hu J. S., Ren L. L., Guo Y. G., Liang H. P., Cao A. M., Wan L. J., Bai C. L.,Angew.Chem.Int.Ed., 2012, 44(8), 1269—1273
[9] Yan H. J., Yang J. H., Ma G. J., Wu G. P., Zong X., Lei Z. B., Shi J. Y., Li C.,J.Catal., 2009, 266(2), 165—168
[10] Huo Y. N., Yang X. L., Zhu J., Li H. X.,Appl.Catal.B:Environ., 2011, 106(1/2), 69—75
[11] Mei Z., Ouyang S., Tang D. M., Kako T., Golberg D., Ye J.,DaltonTrans., 2013, 42(8), 2687—2690
[12] Fan Y. H., Luo Q., Liu G. X., Wang J. X., Dong X. T., Yu W. S., Sun D.,Chin.J.Inorg.Chem., 2014, 30(3), 627—632(范英华, 雒琴, 刘桂霞, 王进贤, 董相廷, 于文生, 孙德. 无机化学学报, 2014, 30(3), 627—632)
[13] Lu Y. H., Wu P. X., Huang J. Y., Chen L. X., Zhu N. W., Dang Z.,Chem.J.ChineseUniversities, 2015, 36(8), 1563—1569(卢勇宏, 吴平宵, 黄俊毅, 陈理想, 朱能武, 党志. 高等学校化学学报, 2015, 36(8), 1563—1569)
[14] Xia S., Lei W., Yang Y. L.,NanoscaleRes.Lett., 2011, 6(40), 562—567
[15] Yong X., Schoonen M. A. A.,Am.Mineral., 2000, 85(3/4), 543—556
[16] Ge L., Han C. C., Xiao X. L., Guo L. L.,Int.J.HydrogenEnergy, 2013, 38(17), 6960—6969
[17] Sun M., Yan T., Yan Q, Liu H. Y., Yan L. G., Zhang Y. F., Du B.,RSCAdv., 2014, 4(38), 19980—19986
[18] Hu S. Z., Li F. Y., Fan Z. P., Wang F., Zhao Y. F., Lv Z. B.,DaltonTrans., 2015, 44(1), 1084—1092
[19] Li D., Wu Z. D., Xing C. S., Jiang D. L., Chen M., Shi W. D., Yuan S. Q.,J.Mol.Catal.A:Chem., 2014, 395(1), 261—268
[20] Chen W., Chen Z. L., Liu T. Y., Jia Z. M., Liu X. H.,J.Environ.Chem.Eng., 2014, 2(3), 1889—1897
[21] Wang D. S., Duan Y. D., Luo Q. Z., Li X. Y., Bao L. L.,Desalination, 2011, 270(1—3), 174—180
[22] Lu M. L., Pei Z. X., Weng S. X., Feng W. H., Fang Z. B., Zheng Z. Y., Huang M. L., Liu P.,Phys.Chem.Chem.Phys., 2014, 16(39), 21280—21288
[23] Sun M., Yan Q., Yan T., Li M. M., Wei D., Wang Z. P., Wei Q., Du B.,RSCAdv., 2014, 4(1), 31019—31027
[24] Liu L. Y., Yang L., Pu Y. T., Xiao D. Q., Zhu J. G.,Mater.Lett., 2012, 66(1), 121—124
[25] Ge L., Han C. C., Liu J.,Appl.Catal.B:Environ., 2011, 108/109(1), 100—107
[26] Joyce S. R., Thirumala R. G., Pushpa M. V., Babu B., Rama K. C., Ravikumar R. V. S. S. N.,J.AlloysComp., 2015, 628(1), 39—45
[27] Kiruthigaa G., Manoharan C., Raju C., Dhanapandian S., Thanikachalam V.,Mater.Sci.Semicon.Proc., 2014, 26(1), 533—539
[29] Ma D. K., Zhou H. Y., Zhang J. H., Qian Y. T.,J.Mater.Chem.Phys., 2008, 111(2/3), 391—395
[30] Liu H., Su Y., Chen P., Wang Y.,J.Mol.Catal.A:Chem., 2013, 378(1), 285—292
[31] Liu H., Jin Z. T., Xu Z. Z.,DaltonTrans., 2015, 44(1), 14368—14375
[32] Dong F., Zhao Z. W., Xiong T., Ni Z. L., Zhang W. D., Sun Y. J., Ho W. K.,ACSAppl.Mater.Interf., 2013, 5(21), 11392—11401
[33] Cao J., Luo B. D., Lin H. L., Xu B. Y., Chen S. F.,J.Hazard.Mater., 2012, 217/218(1), 107—115
[34] Wang Y., Wang X. C., Antonietti M.,Angew.Chem.Int.Ed., 2012, 51(1), 68—89
[35] Ge L., Han C.,Appl.Catal.B:Environ., 2012, 117/118(1), 268—274
[36] Zhu Y. P., Li J., Ma T. Y., Liu Y. P., Du G. H., Yuan Z. Y.,J.Mater.Chem.A, 2014, 2(1), 1093—1101
[37] Zhang K., Kim W. J., Ma M., Shi X. J., Park J. H.,J.Mater.Chem.A, 2015, 3(1), 4803—4810
[38] Li Y. G., Wei X. L., Li H. J., Wang R. R., Feng J., Yun H., Zhou A. N.,RSCAdv., 2015, 5(1), 14074—14080
[39] Liu G., Niu P., Sun C. H., Smith S. C., Chen Z. G., Lu G. Q., Cheng H. M.,J.Am.Chem.Soc., 2010, 132(33), 11642—11648
[40] Jin R. R., You J. G., Zhang Q., Liu D., Hu S. Z., Gui J. Z.,ActaPhys-Chim.Sin., 2014, 30(9), 1706—1712(金瑞瑞, 游继光, 张倩, 刘丹, 胡绍争, 桂建舟. 物理化学学报, 2014, 30(9), 1706—1712)
[41] Zhang J., Wang Y. J., Hu S. Z.,ActaPhys-Chim.Sin., 2015, 31(1), 159—165(张健, 王彦娟, 胡绍争. 物理化学学报, 2015, 31(1), 159—165)
[42] Ji L., Wang H. R., Yu R. M.,Chem.J.ChineseUniversities, 2014, 35(10), 2170—2176(姬磊, 王浩人, 于瑞敏. 高等学校化学学报, 2014, 35(10), 2170—2176)
(Ed.: S, Z, M)
Preparation of Zn0.11Sn0.12Cd0.84S1.12/g-C3N4Heterojunctions and Their Photocatalytic Performance Under Visible Light†
ZHANG Qian, HU Shaozheng*, LI Fayun, FAN Zhiping, WANG Qiong, WANG Fei, LI Wei, LIU Daosheng*
(CollegeofChemistry,ChemicalEngineering,andEnvironmentalEngineering,LiaoningShihuaUniversity,Fushun113001,China)
Visible light responsive ZnSnCdS/g-C3N4heterojunction photocatalysts were synthesizedviaa simple hydrothermal treatment. X-ray diffraction(XRD), scanning electron microscopy(SEM), UV-Vis spectroscopy, Fourier transform infrared spectroscopy(FTIR), inductively coupled plasma-mass spectrometry(ICP-MS), photoluminescence(PL) spectroscopy, and X-ray photoelectron spectroscopy(XPS) were used to characterize the prepared catalysts. The results indicated that the heterojunctions were formed by the formation of C—S bond across the g-C3N4/ZnSnCdS interface, which facilitates interfacial charge transfer and improves the separation efficiency of electron-hole pairs. The activities of catalysts were tested in photocatalytic rhodamine B(RhB) degradation under visible light. The results indicated that the ZnSnCdS/g-C3N4heterojunction photocatalysts show obvious higher photocatalytic activity than the single g-C3N4or ZnSnCdS. With the optimal g-C3N4mass fraction of 20%, the as-prepared heterojunction photocatalyst displays the highest RhB degradation rate, which is 32.3 and 4.9 times that of single g-C3N4and ZnSnCdS, respectively. Not only ZnSnCdS but other ternary metal sulfides, ZnMoCdS, MoNiCdS and NiSnCdS, can combine with g-C3N4to form the heterojunction catalyst to promote the separation rate of electrons-holes.
g-C3N4; Ternary metal sulfide; Heterojunction catalyst; Photocatalysis; Visible light
10.7503/cjcu20150721
2015-09-16.
日期: 2016-01-24.
辽宁省教育厅一般项目(批准号: L2012128)资助.
O644
A
联系人简介:胡绍争, 男, 博士, 副教授, 主要从事新型光催化材料研究. E-mail: hushaozhenglnpu@163.com
刘道胜, 男, 博士, 副教授, 主要从事多孔材料制备及应用研究. E-mail: liudaoshenglnpu@163.com
† Supported by the Education Department of Liaoning Province, China(No. L2012128).
猜你喜欢
科学之友(2022年11期)2022-11-03
大学物理(2022年9期)2022-09-28
工业水处理(2022年6期)2022-06-23
当代水产(2021年3期)2021-07-20
石油化工高等学校学报(2021年3期)2021-07-15
物理通报(2020年7期)2020-07-01
物理化学学报(2017年3期)2017-03-11
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
原子与分子物理学报(2015年3期)2015-11-24
