
Criegee中间体CH3CHOO与OH自由基反应机理的理论研究
2016-12-02 03:03高志芳王渭娜刘峰毅王文亮
高等学校化学学报 2016年3期
高志芳, 王渭娜, 马 倩, 刘峰毅, 王文亮
(陕西省大分子科学重点实验室, 陕西师范大学化学化工学院, 西安 710119)
Criegee中间体CH3CHOO与OH自由基反应机理的理论研究
高志芳, 王渭娜, 马 倩, 刘峰毅, 王文亮
(陕西省大分子科学重点实验室, 陕西师范大学化学化工学院, 西安 710119)
采用CCSD(T)//B3LYP/6-311+G(d,p)方法研究了Criegee中间体CH3CHOO与OH自由基反应的微观机理. 结果表明, 上述反应存在抽氢、 加成-分解和氧化3类反应通道, 其中, syn-CH3CHOO+OH以抽β-H为优势通道, 表观活化能为-4.88 kJ/mol; anti-CH3CHOO+OH则以加成-分解反应为优势通道, 表观活化能为-13.25 kJ/mol. 在加成-分解和氧化反应通道中, anti-构象的能垒均低于syn-构象, 而抽氢反应则是syn-(β-H)的能垒低于anti-构象. 速率常数计算表明, anti-构象的加成-分解反应通道具有显著的负温度效应; syn-和anti-构象的氧化通道具有显著的正温度效应. 3类反应具有显著不同的温度效应, 说明通过改变温度可显著调节3类反应的相对速率.
Criegee中间体; CH3CHOO自由基; OH自由基; 反应机理; 速率常数
人们的日常生活及工业燃烧显著增加了城市地区大气中短链烯烃(如, 乙烯、 丙烯和丁烯等)的含量[1], 这些不饱和碳氢化合物是大气中的主要污染物之一, 约占挥发性有机化合物的13%[2]. 含4个或4个以下C原子的烯烃被大气中的O3氧化生成活性较高的Criegee中间体(CIs) R2COO, 它们既能发生单分子异构化反应[3], 又可与大气物种CO[4], H2O[5,6], SO2[7]及HOx[8]等发生双分子反应. Anglada等[9]报道, CH2OO与OH反应首先生成H2CO和HO2, 其中, HO2可以引起CIs的聚合, 生成二次有机气溶胶(Secondary organic aerosol, SOA). 文献[10]也报道, CIs与大气物种反应是SOA形成的主要途径之一. CH3CHOO是最简单Criegee中间体CH2OO的甲基取代物, 其形成机理如Scheme 1所示. O3的两端氧首先协同加到烯烃双键碳上, 形成分子臭氧化物(POZ), 然后发生C—C键断裂分解形成羰基化合物(1)和CIs(2)[3]. 此过程是强放热反应, 过剩的能量可促使syn-CH3CHOO通过β-H迁移-分解通道生成CH2CHO+OH[11]. 文献[12]报道, anti-CH3CHOO通过分子内的插氧反应(端位O原子插入到α-C-H之间)异构生成CH3C(O)OH, 并分解产生OH自由基. 因此, CIs既是大气HOx的重要来源[11], 同时也是消耗大气HOx的主要途径. CIs与大气物种的反应对大气HOx浓度与循环、 气溶胶形成及气候变化均有重要影响[13,14], 已成为大气化学研究中的一个热点课题. 但是目前关于CIs的研究主要集中在其电子结构、 光谱性质以及CH2OO的反应机理上, 未见CH3CHOO与OH反应的详细机理、 产物分布及优势通道速率常数等信息的报道. 本文采用CCSD(T)//B3LYP/6-311+G(d,p)双水平方法研究了CH3CHOO+OH反应的微观机理, 以期为实验研究提供理论指导.
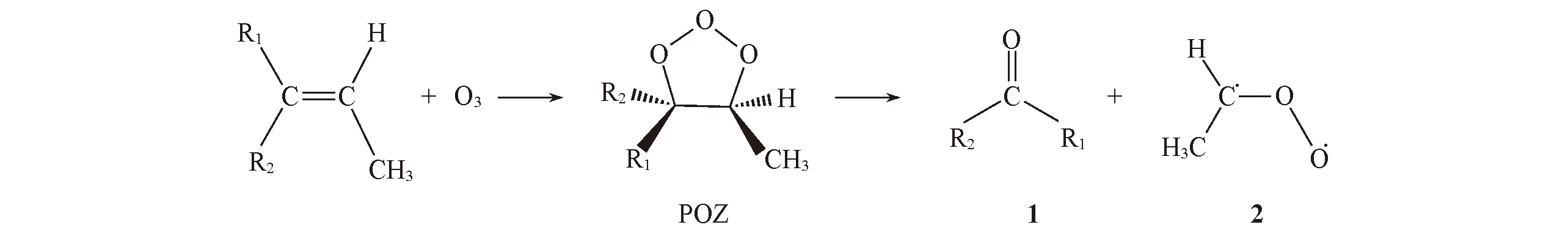
Scheme 1 Ozonolysis of alkenes forming carbonyl compound and CH3CHOO
1 计算方法
文献[15,16]报道, syn-CH3CHOO与anti-CH3CHOO构象的基态电子结构均为闭壳层单重态. 本文选用B3LYP/6-311+G(d,p)方法对反应通道上的各个驻点(反应物、 中间体、 过渡态和产物)的几何构型进行全参数优化, 并进行内禀反应坐标(IRC)分析, 以证实反应物、 中间体、 过渡态及产物的相关性. 基于频率振动分析确认各驻点的性质. 为了更加精确地预测具有最小能垒的反应路径信息, 采用CCSD(T)方法进行了单点能计算, 得到各反应通道中物种的能量信息与反应能垒. 所有计算均采用Gaussian 09程序包[17]完成.
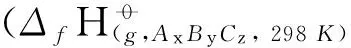
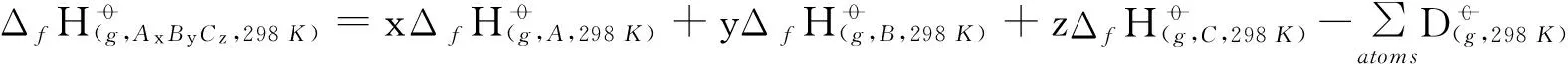
(1)
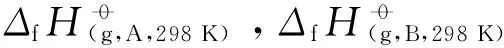
(2)
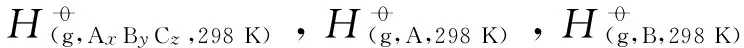
(3)
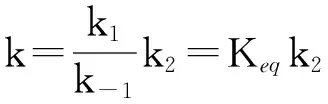
(4)
(5)
其中, 配分函数QCH3CHOO,QOH及QRC由Zhangrate程序计算所得;ER和ERC分别为CCSD(T)//B3LYP/6-311+G(d,p)水平下反应物及中间体的能量;σ为对称数.
2 结果与讨论
2.1 syn-/anti-CH3CHOO构象及旋转能垒
syn-/anti-CH3CHOO的几何优化构象如图1所示. 从图1可见, 采用B3LYP与CCSD(T)方法优化得到的主要键长数据非常接近, 且T1诊断值均小于0.044, 双重态S2最大值不超过0.800, 消除自旋污染后与无污染本征值0.750 完全一致[表S1(见本文支持信息)], 说明波函数自旋污染并不严重. 采用同种计算方法优化得到的2种构象中主要键长非常接近, 但2种构象的能量差为14.08 kJ/mol, syn-构象更稳定. 这与文献[21]报道的syn-构象比anti-构象大约低15.06 kJ/mol的结论一致. syn-构象稳定的原因是—CH3上的2个H与端O之间距离较小(0.262 nm), 形成较弱的分子内氢键. 对比syn-CH3CHOO与anti-CH3CHOO结构可知, syn-与anti-之间的转化必须通过COO单元的CO键的旋转来实现. 计算结果表明, CH3CHOO中C—O键键长为0.126~0.127 nm, 远小于C—O单键键长(约0.14 nm), 稍大于醛或酮类化合物CO双键键长(~0.121 nm). 说明CH3CHOO中C—O具有双键性质特征, 旋转异构化的能垒必然很高. CCSD(T)计算结果证实, syn-CH3CHOO异构化为anti-CH3CHOO的能垒高达173.08 kJ/mol.可以推测, 只有在极端条件下才可能促使旋转异构化发生. 室温下, anti-CH3CHOO与 syn-CH3CHOO几乎就是2个独立物种, syn-CH3CHOO为主要成分. 实验上已经由紫外光谱及光电离技术证实了这一结论[22~25]. 此外, 标题反应中生成了一系列含过氧自由基的化(复)合物, 部分产物复合物PC处于较深的势井中, 表明这些产物复合物具有较高稳定性. 有些反应复合物RC或过氧自由基产物能量较高不稳定. 目前, 关于此类过氧自由基复合物的生成焓数据报道甚少. 本文采用原子化方法计算了部分物种的生成焓, 结果列于表1. 结果表明, 对于一些稳定分子采用G4方法计算的生成焓值与实验值非常接近, 说明所选计算方法是合理的. 产物复合物PC的生成焓为负值, 稳定性较高. 而反应物CH3CHOO、 抽氢产物CH3COO和CH2CHOO、 反应复合物RC的生成焓为正值, 说明这些物种在大气条件下是不稳定的, 这也可能是聚合形成SOA的原因之一.
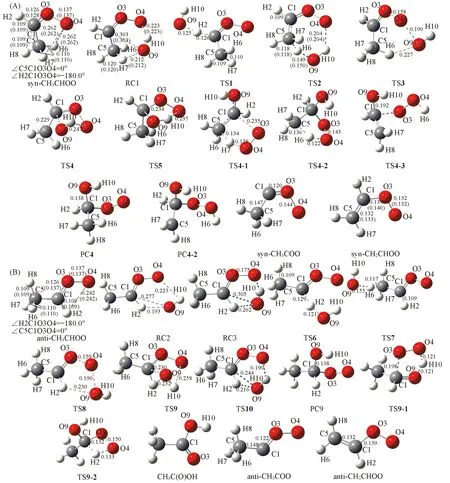
Fig.1 Optimized geometries of stationary point of syn-CH3CHOO+OH(A) and anti-CH3CHOO+OH(B) at the B3LYP/6-311+G(d,p) levelThe values in parentheses are at the CCSD(T)/6-311+G(d,p) level. The bond lengths are in nm.

Table 1 Formation enthalpies ΔfH0— for some species at the G4 level
2.2 CH3CHOO+OH反应机理
参照文献[27,28]方法, 分别对CH3CHOO单/三重态结构进行结构优化和能量计算, 并分别研究了CH3CHOO与OH在双/四重态上的优势反应通道. 结果发现, 三重态syn-CH3CHOO能量比单重态高 127.03 kJ/mol, 三重态anti-CH3CHOO能量比单重态能量高114.90 kJ/mol. 所以在大气条件下, 单重态CH3CHOO的稳定性远大于三重态. 对于CH3CHOO与OH的反应, 无论抽氢反应还是加成反应, 低自旋态通道反应的能垒均比高自旋态通道的能垒低, 说明低自旋态通道为绝对优势通道[图S1(见本文支持信息)]. 因此, Scheme 2仅示出CH3CHOO+OH反应体系在二重态势能面上所有可能的通道, 主要包括抽氢、 加成-分解和氧化3种反应类型. 图2~图4分别给出了各类反应的势能面.
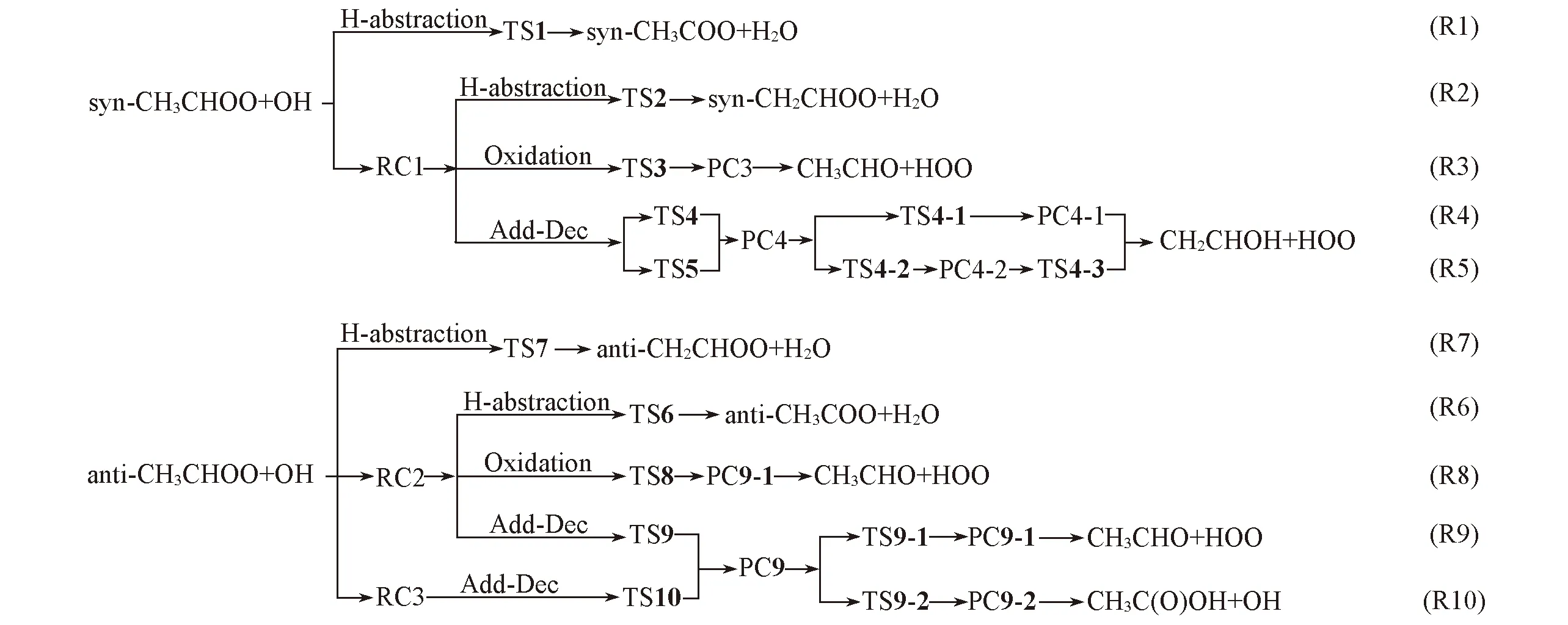
Scheme 2 Possible reaction channels of CH3CHOO with OH radical
2.2.1 抽氢反应 抽氢反应包括OH抽取syn-/anti-CH3CHOO上的α-H(通道R1与R6)和β-H. 由于syn-构象中β-C上H6和H7均与端O存在弱的分子内相互作用形成弱氢键, 与β-C上H8可能存在差异, 因此—CH3上的3个H可分为两类. 但选用B3LYP, M06-2X, CCSD(T)和QCISD等多种方法均未能找到OH分别抽取syn-和anti-中H8的过渡态. 分别以syn-/anti-的C5—H8键长为变量进行能量计算, 得到了syn-/anti-结构的能量随键长变化的刚性扫描图[图S2(见本文支持信息)]. 若将最高点与稳定构型的能量差作为活化能, 则从扫描图可看出, OH抽取 syn-/anti-上H8的活化能远远高于抽取β-H6(通道R2与R7)的能垒. 因此, 仅选择抽取β-H6(通道R2与R7)为代表讨论OH抽取β-H的机理.

Fig.2 Potential energy surface of the hydrogen abstraction at the CCSD(T)// B3LYP/6-311+G(d,p) level
从图2可以看出, OH抽取syn-构象的α-H(通道R1)属于直接抽氢, 并不形成复合物, 是C—H键断裂和O—H键形成同时进行的过程. 经过渡态TS1生成syn-CH3COO+H2O, 此过程能垒为120.49 kJ/mol, 放热33.28 kJ/mol; syn-构象的β-H与OH(R2)属于间接抽氢方式, 即首先形成一个七元环的氢键复合物RC1, 结合能为-16.62 kJ/mol. 从RC1出发, 经过渡态TS2生成过氧自由基syn-CH2CHOO和H2O, 这一过程伴随显著的放热效应(ΔE=-197.15 kJ/mol). 在syn-CH2CHOO中, C—C键长从反应物的0.147 nm显著缩短到0.132 nm, 与烯烃中的C—C键长接近, C—O键长从0.126 nm拉长到0.139 nm, 接近醚类的C—O键长. 此基元过程能垒仅为11.74 kJ/mol , 表观能垒为很小的负值. 因此, 预测在一定温度范围内, 速率常数具有弱的负温度效应. 显然, 与直接抽α-H相比(R1), 从热力学与动力学的角度分析, 抽取β-H(R2)反应是一个自发的快速反应过程. 上述2种不同的抽氢方式对应过渡态能垒高度TS1(α-H)远大于TS2(β-H), 这可能与环状过渡态的形成有关, TS2是七元环的构型, 是张力较小的稳定结构. 因此, 对于syn-CH3CHOO, OH抽取β-H比抽取α-H容易得多.
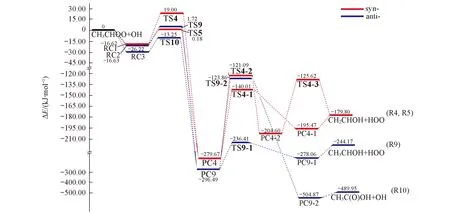
Fig.3 Potential energy surface for addition-decomposition mechanism at the CCSD(T)//B3LYP/6-311+G(d,p) level
anti-构象抽氢过程与syn-构象抽氢过程不同, OH抽取α-H(R6) 属于间接抽氢方式, 首先形成一个六元环的氢键复合物RC2, 结合能为-16.63 kJ/mol, 经过渡态TS6需克服27.30 kJ/mol的基元能垒, 生成anti-CH3COO+H2O. 此过程的表观活化能为10.67 kJ/mol; OH抽取anti-构象β-H(通道R7)为直接抽氢机理, 经过渡态TS7需克服43.25 kJ/mol的能垒, 生成anti-CH2CHOO+H2O, 放热183.40 kJ/mol. 因此, 从动力学方面考虑, 与syn-CH3CHOO中抽取β-H远易于α-H的结论不同, anti-CH3CHOO中是抽取α-H远易于抽取β-H. 综合分析syn-/anti-CH3CHOO的4条抽氢通道(R1, R2, R6, R7)可以得出, 直接抽氢方式的能垒总是大于间接抽氢方式, 即间接抽氢方式优于直接抽氢. 在对CH2OO+OH[9]及CH3CH2O+HCHO[29]体系的研究中也有类似.
2.2.2 加成-分解反应 加成-分解反应是指OH上的O先加成到α-C上生成反应复合物RC, 经过渡态生成产物复合物PC, PC再分解释放出HOO或OH. 此处应特别强调的是分解产生HOO或OH中的O全部来自CH3CHOO. syn-/anti-构象的加成-分解反应包括通道R4和R5及R9和R10.
从图1与图3可知, 在OH与syn-CH3CHOO的加成反应中, 首先形成一个五元环氢键复合物RC1, 从RC1出发, 分别经过渡态TS4与TS5形成同一产物复合物PC4, 同时放出大量的热(ΔE=-279.67 kJ/mol), 同时促使2个异构化反应继续进行, 即分别经过渡态TS4-1与TS4-2形成相同产物CH2CHOH+HOO. TS4与TS5的区别在于OH中H的取向不同, TS5中羟基H指向端O4, 形成一个五元环结构. TS4中羟基H6并未指向端O4, 没有形成环状结构, 导致TS4的能垒比TS5高18.82 kJ/mol. 因此, 加成-分解以R5通道为主要方式进行.另外, syn-CH3CHOO的抽β-H通道R2与加成-分解R5通道均具有较低的基元能垒和近似为零(或负)的表观活化能, 因此这2个通道均是快速过程.
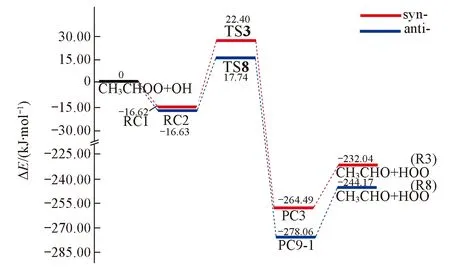
Fig.4 Potential energy surface for oxidation mechanism at the CCSD(T)//B3LYP/6-311+G(d,p) level
与syn-CH3CHOO的加成-分解不同, anti-CH3CHOO先与OH形成2个结构相似的复合物RC2与RC3, 分别经过渡态TS9与TS10形成同一产物复合物PC9, 然后经过渡态TS9-1生成产物CH3CHO+HOO, 或经过渡态TS9-2生成产物CH3C(O)OH+OH. 显然, 与syn-CH3CHOO加成-分解生成产物(CH2CHOH+HOO)不同, 最大差异是anti-CH3CHOO加成-分解中有CH3COOH+OH生成, 且此羟基并非原反应物OH, 而是发生了OH交换反应, 原反应物OH加到α-C上, 产物OH则来自anti-CH3CHOO的端O与α-H. 另外, 虽然加成-分解中生成的HOO中2个氧原子均来自Criegee中间体, 但在syn-加成-分解反应中, H原子来自β-H, 而anti-加成-分解反应中, H原子来自反应物OH上的H. 从图3还可见, anti-CH3CHOO加成-分解生成CH3CHO+HOO通道R9的多个步骤均具有负的表观活化能, 在动力学上更有利, 是标题反应的最优通道. anti-CH3CHOO加成-分解生成CH3C(O)OH+OH通道(R10)虽然从PC9到PC9-2步骤具有较高的基元能垒, 但表观活化能仍为负值, 且放热高达-489.95 kJ/mol, 也应是优势通道之一.
2.2.3 氧化反应通道R3与R8 氧化反应也称抽氧反应, 与加成-分解反应不同之处是产物HOO中仅端O来自于Criegee中间体, 也即OH抽取CIs端位上的O原子, 相当于CIs充当氧化剂, 端位上的O原子迁移到OH上, 形成过氧自由基HOO.
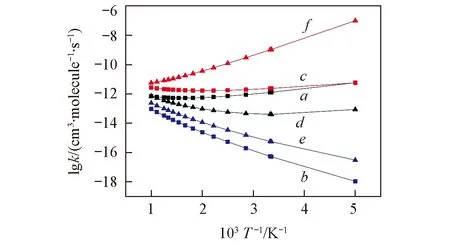
Fig.5 Fitted plots of the rate constants vs. the reciprocal of temperature for the favorable channels over the range of 200—1000 Ka. syn-β-Hab(R2); b. syn-Oxi(R3); c. syn-Add(R5); d. anti-α-Hab(R6); e. anti-Oxi(R8); f. anti-Add(R10).
从图4可见, anti-+OH的反应能垒稍低于syn-+OH反应, 且多放热12.13 kJ/mol. 因此可以推断, anti-CH3CHOO氧化OH生成HOO的活性稍高于syn-CH3CHOO. Thompson等[4]报道了15种不同取代的Criegee中间体氧化CO生成CO2的反应, 证明由于取代基空间位阻作用, anti-构象的氧化活性大于syn-构象. 特别指出的是, 氧化反应与加成-分解反应均生成了HOO, 但反应机理显著不同, 氧化反应是CH3CHOO端位的一个O原子迁移到OH上生成HOO, 即只有端氧来自CIs; 而加成-分解反应生成HOO的2个氧原子均来自CIs. 可用同位素标定方法检测其2种反应机理的最终产物, 验证本文所提机理的合理性.
2.3 优势通道速率常数
为了便于比较, 在抽氢、 加成-分解和氧化3类反应中各选一个优势反应通道, 即syn-CH3CHOO+OH体系中的R2, R3与R5及anti-CH3CHOO+OH体系中的R6, R8与R10, 基于传统过渡态理论计算其反应速率常数, 结果列于表S2(见本文支持信息)和图5. 由图5可知, anti-构象的加成-分解反应(R10)速率常数最大, 300 K时达到1.03×10-9cm3·molecule-1·s-1, 且具有显著的负温度效应. 这与Taatjes等[30]报道的anti-CH3CHOO与H2O的加成反应活性高于syn-构象的实验结论相吻合.
从图5还可看出, syn-构象的抽β-H(R2)、 加成-分解反应(R5)速率常数随温度变化很小. 与几乎为零的表观活化能相对应, 在低温范围具有微弱的负温度效应, 高温范围具有微弱的正温度效应; 3类反应中氧化反应(通道R3, R8)速率常数最小, 300 K时分别为5.51×10-17和5.79×10-16cm3·molecule-1·s-1, 且具有显著的正温度效应, 与正的表观活化能(分别为22.40和17.74 kJ/mol)相对应. 按反应类型比较, 3类反应对应的表观活化能存在显著差异, 反应活性明显不同. 加成-分解反应活性最高, 氧化反应活性最小, 抽氢反应活性居中, 且3类反应具有显著不同的温度系数, 说明改变温度可显著调节此3类反应的相对速率; 对比syn-与anti-构象, anti-构象的加成-分解及氧化反应活性大于syn-构象, 而抽氢反应活性则是syn-构象大于anti-构象.
3 结 论
在CCSD(T)//B3LYP/6-311+G(d,p)双水平上对CH3CHOO+OH的反应机理进行了研究, 结果表明, 标题反应包括抽氢、 加成-分解和氧化3类反应通道, syn-构象与OH反应以抽β-H反应生成syn-CH2CHOO+H2O为优势通道, anti-构象与OH反应以加成-分解反应产生成CH3CHO+HOO为优势通道, 且均是低基元能垒、 负表观活化能的放热反应通道. 计算的速率常数表明, 3类反应表现为不同的温度效应. anti-构象加成-分解反应通道具有显著的负温度效应, syn-构象抽β-H反应低温范围表现出微弱负温度效应, 高温范围表现出微弱的正温度效应, syn-/anti-构象的氧化通道具有显著的正温度效应; anti-构象反应的加成-分解和氧化反应速率大于syn-构象反应的速率, 而抽氢反应速率则是syn-构象大于anti-构象. CH3CHOO是对流层大气化学污染的重要参与者, 与OH自由基所发生的抽氢、 加成-分解和氧化3类反应具有显著不同的温度系数. 因此, 可以通过改变温度显著调节3类反应的相对速率, 以改变CH3CHOO+OH反应产物在大气中的分布.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20150742.
[1] Paulson S. E., Chung M. Y., Hasson A. S.,J.Phys.Chem.A, 1999, 103(41), 8125—8138
[2] Jacobson M.,AtmosphericEnvironment, 1997, 31(3), 511—512
[3] Johnson D., Marston G.,Chem.Soc.Rev., 2008, 37(4), 699—716
[4] Kumar M., Busch D. H., Subramaniam B., Thompson W. H.,J.Phys.Chem.A, 2014, 118(10), 1887—1894
[5] Ryzhkov A. B., Ariya P. A.,Phys.Chem.Chem.Phys., 2004, 6(21), 5042—5050
[6] Qi B., Chao Y. T.,ActaChim.Sinica, 2007, 65(19), 2117—2123(齐斌, 晁余涛. 化学学报, 2007, 65(19), 2117—2123)
[7] Jiang L., Xu Y. S., Ding A. Z.,J.Phys.Chem.A, 2010, 114(47), 12452—12461
[8] Bo L., Tan X. F., Long Z. W., Wang Y. B., Ren D. S., Zhang W. J.,J.Phys.Chem.A, 2013, 3(8), 1693—1699
[9] Mansergas A., Anglada J. M.,J.Phys.Chem.A, 2006, 110(11), 4001—4011
[10] Sadezky A., Winterhalter R., Kanawati B., Rompp A., Spengler B., Mellouki A., Bras G. L., Chaimbault P., Moortgat G. K.,Atmos.Chem.Phys., 2008, 8(10), 2667—2699
[11] Kumar M., Busch D. H., Subramaniam B., Thompson W. H.,Phys.Chem.Chem.Phys., 2014, 16, 22968—22973
[12] Nguyen T. N., Putikam R., Lin M. C.,J.Chem.Phys., 2015, 142(12), 124312
[13] Lee S., Kamens R. M.,Atmos.Environ., 2005, 39(36), 6822—6832
[14] Heard D. E., Carpenter L. J., Creasey D. J., Hopkins J. R., Lee J. D., Lewis A. C., Pilling M. J., Seakins P. W., Carslaw N., Emmerson K. M.,Geo.Res.Lett., 2004, 31(18), 355—366
[15] Li H.W., Fang Y., Kidwell N. M., Beames J. M., Lester M. I.,J.Phys.Chem.A, 2015, 119(30), 8328—8337
[16] Kettner M., Karton A., McKinley A. J., Wild D. A.,ChemicalPhysicsLetters, 2015, 621, 193—198
[17] Frisch M. J., T. G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A.G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A.,Gaussian09, Gaussian Inc., Wallingford CT, 2009
[18] Curtiss L. A., Raghavachari K., Redfern P. C., Pople J. A.,J.Chem.Phys., 1997, 106, 1063—1079
[19] Curtiss L. A., Redfern P. C., Raghavachari K.,J.Chem.Phys., 2007, 127(12), 66—70
[20] Zhang S. W., Truong N. T.,VKLabVersion1.0, University of Utah, Salt Lake City, 2001
[21] Kuwata K. T., Hermes M. R., Carlson M. J., Zogg C. K.,J.Phys.Chem.A, 2010, 114(34), 9192—9204
[22] Nakajima M., Yue Q., Endo Y.,JournalofMolecularSpectroscopy, 2015, 310, 109—112
[23] Smith M. C., Ting W. L., Chang C. H., Takahashi K., Boering K. A., Lin J. J. M.,J.Chem.Phys., 2014, 141(7), 074302
[24] Nakajima M., Endo Y.,J.Chem.Phys., 2014, 140(1), 011101
[25] Lin H. Y., Huang Y. H., Wang X., Bowman J. M., Nishimura Y., Witek H. A., Lee Y. P.,Nat.Commun., 2015, 6, 7012
[26] NIST Chemistry Webbook(http://webbook.nist.gov/chemistry)
[27] Li D. M., Wang Y., Han K. L.,Coord.Chem.Rev., 2012, 256, 1137—1150
[28] Balucani N., Capozza G., Leonori F., Segoloni E., Casavecchia P.,Int.Rev.Phys.Chem., 2006, 25, 109—163
[29] Guo S., Wang W. N., Jin L. X., Wang S., Wang W. L.,Chem.J.ChineseUniversities, 2014, 36(8), 1300—1306(郭莎, 王渭娜, 靳玲侠, 王帅, 王文亮. 高等学校化学学报, 2014, 36(8), 1300—1306)
[30] Taatjes C. A., Welz O., Eskola A. J., Savee J. D., Scheer A. M., Shallcross D. E., Rotavera B., Lee E. P. F., Dyke J. M., Mok D. K. W., Osborn D. L., Percival C. J.,Science, 2013, 340(6129), 177—180
(Ed.: Y, Z)
† Supported by the National Natural Science Foundation of China(Nos.21473108, 21473107) and the Innovative Team of Key Science and Technology of Shaanxi Province, China(No.2013KCT-17).
Theoretical Studies on the Reaction Mechanism of Criegee Intermediates CH3CHOO with OH Radicals†
GAO Zhifang, WANG Weina, MA Qian, LIU Fengyi, WANG Wenliang*
(SchoolofChemistryandChemicalEngineering,ShaanxiNormalUniversity,KeyLaboratoryforMacromolecularScienceofShaanxiProvince,Xi’an710119,China)
The reaction mechanism of CH3CHOO with OH was studied at the CCSD(T)//B3LYP/6-311+G(d,p) level. The results show that the detailed mechanism mainly includes hydrogen abstraction, addition-decomposition and oxidation reactions. The calculations show thatβ-H abstraction reaction is favoured in the syn-CH3CHOO reaction with OH with a apparent activation energy(Eapp) of -4.88 kJ/mol, whereas the addition-decomposition channel is advantaged in the reaction of anti-CH3CHOO with OH in combination with aEappof -13.25 kJ/mol. The barriers of anti-CH3CHOO are lower than that of the syn-CH3CHOO in addition-decomposition and oxidation reactions, while the syn-CH3CHOO is lower than anti-CH3CHOO in theβ-H abstraction pathway. The rate constant of addition-decomposition in the reaction of anti-CH3CHOO satisfies a strong negative temperature dependent, while oxidation channels of syn-/anti-CH3CHOO obey a positive temperature effect. The temperature dependent of three kind of reactions are different, which makes their relative rate can be controlled by adjusting the temperature.
Criegee intermediate; CH3CHOO radical; OH radical; Reaction mechanism; Rate constant
10.7503/cjcu20150742
2015-09-22.
日期: 2016-01-24.
国家自然科学基金(批准号: 21473108, 21473107)和陕西省重点科技创新团队基金(批准号: 2013KCT-17)资助.
O643.1
A
联系人简介: 王文亮, 男, 教授, 博士生导师, 主要从事理论与计算化学研究. E-mail: wlwang@snnu.edu.cn
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
唐山师范学院学报(2020年6期)2020-04-16
唐山师范学院学报(2019年3期)2019-06-18
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
温州大学学报(自然科学版)(2016年1期)2016-10-27
科技视界(2016年24期)2016-10-11
