
p53突变体Y220C小分子稳定剂的虚拟筛选
2016-12-01 07:21:22丁吉勇沈洪辰刘夫锋
高等学校化学学报 2016年4期
丁吉勇, 沈洪辰, 刘夫锋
(天津大学化工学院生物工程系, 系统生物工程教育部重点实验室, 天津 300072)
p53突变体Y220C小分子稳定剂的虚拟筛选
丁吉勇, 沈洪辰, 刘夫锋
(天津大学化工学院生物工程系, 系统生物工程教育部重点实验室, 天津 300072)
为了获得p53突变体的稳定剂, 依次利用利宾斯基五原则, 通过2次分子对接和全原子分子动力学(MD)模拟从DrugBank 4.0 数据库中筛选获得了潜在的稳定剂他克林. 利用MD模拟进一步验证他克林和目标蛋白质之间的亲和作用. 结果表明, 他克林能够紧密结合到Y220C突变所形成的疏水空腔之中; 他克林和目标蛋白质之间的主要作用力为疏水和静电相互作用, 其中疏水相互作用占主导地位. 此外, 他克林分别与目标蛋白质的残基Leu145, Val147和Asp228形成3个氢键. 基于MD模拟轨迹分析了他克林与p53C-Y220C的结合过程. 由硫黄素T荧光光谱进一步证明他克林能够提高p53C-Y220C突变体的稳定性.
癌症; 蛋白质稳定剂; 分子对接; 分子动力学模拟; p53; 他克林
p53是一种重要的肿瘤抑制蛋白质, 具有阻滞肿瘤细胞生长、 促进肿瘤细胞凋亡以及抑制肿瘤血管生成等多种生物学功能[1]. p53含有5个结构域, 分别为N末端反式激活结构域、 脯氨酸富集域、 核心DNA结合域、 四聚体区域和C末端区域. p53共含有393个氨基酸, 其中94~292位氨基酸为该蛋白质的核心结构域, 被定义为p53C. 据统计, 约50%的人类癌症是由p53突变引起的[2,3]. 其中大多数突变位于p53C结构域, 如Y220C, R248Q, R248W, R273H, R273C, R175H, G245S, R249S 和R282H(详细信息可查阅数据库http: //www-p53.iarc.fr). 当然, 除了上述常见的突变位点外, 在p53的C或N末端结构域也会发生一些突变, 如R337H[4].
野生型p53C的稳定性很差, 其熔解温度约为44 ℃, 在体温环境下该蛋白质的半衰期约为9 min[5]. 而突变会进一步降低p53C的稳定性, 加速其变性及聚集沉淀, 从而阻碍其抑癌功能的发挥, 同时还会大大增强肿瘤的转移和侵袭能力. 因此, 开发能够提高p53突变体稳定性的药物是一个有效的治疗癌症方法. 但大多数p53的突变位于DNA结合域, 稳定剂结合到该部位可能会阻碍p53与DNA结合, 从而严重影响p53的抑癌功能. 据世界卫生组织统计, 在最易引发癌症的p53突变体中, Y220C突变体排在第9位. 全世界每年约7.5万例癌症是由Y220C突变所引起的[6]. 因此, 设计p53C-Y220C突变体的稳定剂迫在眉睫. p53C-Y220C突变体的二级结构见图S1(见本文支持信息). 位于S3/S4环和S7/S8环之间的220位酪氨酸突变成半胱氨酸会在p53表面形成一个疏水空腔[本文支持信息中图S2(B)所示], 而Cys220位于该空腔底部. 研究表明该突变会大大降低p53的稳定性, 从而引发p53的变性和聚集沉淀. Wang等[7]发现p53C-Y220C聚集形成淀粉样纤维可以结合硫黄素T(ThT)分子而产生荧光. 另外, Y220C突变所形成的疏水腔远离DNA结合功能结构域, 因此Y220C突变体所形成的疏水性口袋就成为最理想稳定剂的作用位点. 即基于此疏水腔设计获得的稳定剂不会影响p53与DNA的结合, 也就不会影响其抑癌能力.
目前, 已开发出多种分子(有机小分子[8,9]和短肽[10]等)来提高p53C突变体的稳定性. Boeckler等[11]利用虚拟筛选技术从Zinc数据库中筛选获得了与p53C突变体Y220C具有较高亲和力的PhiKan083分子, 该分子能够大大提高该蛋白质的稳定性. Wassman等[12]利用分子动力学(MD)模拟发现了瞬时开关的L1/L3结合位点, 基于该结合位点筛选获得了p53突变体R175H的稳定剂斑点酸, 结果表明该分子能够激活p53的抑癌功能, 从而能够有效抑制人骨肉癌的发生和发展. 虽然已有多种分子被开发用于提高p53C突变体的稳定性, 但至今仍无有效的稳定剂可用于癌症的治疗. 因此亟需开发新型的小分子来提高p53C的稳定性.
基于结构的药物分子筛选需要构建或选择现有的数据库, DrugBank 4.0是收录FDA批准和临床研究阶段的药物分子的数据库[13]. 从该数据库筛选获得的稳定剂分子均可直接作为药物用于p53突变所引起的癌症治疗. 因此, 为了获得能够提高p53C-Y220C热稳定性的药物分子, 本文选择DrugBank 4.0数据库中的小分子, 根据利宾斯基五原则筛选, 利用2次分子对接虚拟筛选找到能够结合到p53C-Y220C疏水腔中的小分子. 然后利用MD模拟验证了这些小分子与目标蛋白之间的亲和力. 分析了他克林和p53C-Y220C突变体之间的亲和作用机理以及他克林稳定p53C-Y220C的作用过程. 利用硫黄素T(ThT)荧光光谱验证了他克林对突变体p53C-Y220C的稳定作用.
1 计算方法
1.1 分子对接
p53C-Y220C(PDB ID: 2J1X)的初始结构选自蛋白质数据库(Protein data bank, PDB)(http: //www.rcsb.org/pdb/)[6]. 利用AutoDockTools 1.5.6 软件对p53C-Y220C分别添加氢原子和Kollman电荷. 在分子对接之前, 首先利用DrugBank 4.0数据库提供的利宾斯基五原则(Lipinski’s rule of five)进行筛选. 然后利用AutoDockTools 1.5.6软件给这些小分子添加Gasteiger电荷后转化为用于AutoDock Vina分子对接所需的pdbqt格式文件[14]. 利用AutoDock Vina 1.1.2程序对小分子与目标蛋白质进行第一次分子对接. 小分子和目标蛋白质之间的亲和作用力通过EVINA计算, EVINA绝对值越大表明它们之间的亲和力越大.
为了进一步降低利用分子对接进行虚拟筛选的误差, 本文利用另一个常用的分子对接软件FlexX进行了第二次分子对接, FlexX软件采用片段生长算法, 它充分考虑了小分子配基的柔性, 将蛋白质看成刚性[15]. 然后利用SYBYL6.92软件中5种打分函数F_Score, ChemScore, D_Score, G_Score和PMF_Score来评价配基与目标蛋白质之间亲和作用力的大小, 其余均采用FlexX的默认值.
1.2 分子动力学模拟
分子动力学(MD)模拟过程中用到的小分子Gromos96力场结构参数利用PRODRG2服务器网站(http: //davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg)构建[16]. 基于Gromos96全原子力场修正小分子的力场参数[17]. 所有的MD模拟均利用GROMACS 4.5软件进行[18]. 水分子选用SPC模型. p53C-Y220C与小分子复合物的MD模拟分别在310 K或 317 K下进行. 利用蛙跳算法对每一步的运动方程积分求解, 积分步长为2 fs. 采用PME方法计算长程静电相互作用[19], 傅里叶格点间距为0.12 nm, 库仑截断距离为0.9 nm, 非键作用原子列表每4个步长更新1次. 短程范德华作用力的截断值为1.4 nm. 利用LINCS算法限制所有化学键[20]. 模拟体系中各原子的初速度利用Maxwell分布随机指定. 首先将小分子-蛋白质复合物置于一个具备周期性边界条件的正方体盒子(8 nm×8 nm×8 nm)中, 然后在盒子中随机地加入水分子, 随后加入5个Cl-代替等量的水分子使模拟体系处于一个中性环境. 经过1000步的能量最小化之后, 利用Berendsen弱耦合方法[21]分别在正则系综(NVT)和等温-等压系综(NPT)下进行200 ps的MD模拟来平衡模拟体系. 最后, 在NPT系综下进行100 ns的MD模拟, 模拟系统的温度和压力分别利用 V-rescale[22]和Berendsen[21]算法控制. 在计算过程中, 每隔2 ps存储一次各原子的坐标, 共存储50000个轨迹进行后续分析. 所有MD模拟计算均在曙光TC2600刀片服务器集群上完成.
1.3 数据分析方法
通过GROMACS 4.5软件包中的一些辅助程序分析模拟轨迹. 配基与目标蛋白质之间的能量采用GROMACS 4.5自带命令g_energy进行分析, g_mindist命令计算指定分子与特定距离之内的残基之间的接触数和该分子与目标蛋白质之间的距离. 根据p53C-Y220C和小分子复合物之间的势能大小来选择最佳构象, 并使用视觉分子动力学(VMD)软件[23](http: //www. ks.uiuc.edu/Research/vmd/) 绘制复合物的构象图.
1.4 p53C-Y220C蛋白质的表达和纯化
含有p53C-Y220C基因片段的重组质粒由金唯智公司提供, 将该质粒转化到大肠杆菌BL21(DE3) 中获得能够进行外源表达p53C-Y220C的基因重组菌, 然后在37 ℃, 220 r/min转速下培养至菌液的OD600值为1.2左右, 然后加入1 mmol/L异丙基-β-D-硫代半乳糖苷, 在25 ℃下继续诱导, 获得目标蛋白质的发酵液. 然后在4 ℃, 220 r/min转速下离心30 min获得菌体, 破碎后离心收集细胞上清液, 分别利用阳离子交换和肝素亲和色谱分离获得高纯度的p53C-Y220C蛋白质用于ThT荧光光谱实验.
1.5 硫黄素T荧光光谱实验
使用20 mmol/L Tris-HCl(pH 7.2)缓冲溶液配制浓度为20 μmol/L的ThT溶液. 分别取200 μL 培养好的含与不含他克林的p53C-Y220C蛋白质培养液(蛋白质浓度为5 μmol/L), 加入2 mL 20 μmol/L ThT溶液, 混合, 然后在Infinite M200 PRO多功能酶标仪上检测溶液的ThT荧光值. 激发波长为440 nm, 发射波长为480 nm. 每个样品扫描3次, 将每次所得扫描数据减去不含目标蛋白质的缓冲液背景后作为实验结果, 计算平均值.
2 结果与讨论
2.1 p53C突变体Y220C稳定剂的虚拟筛选
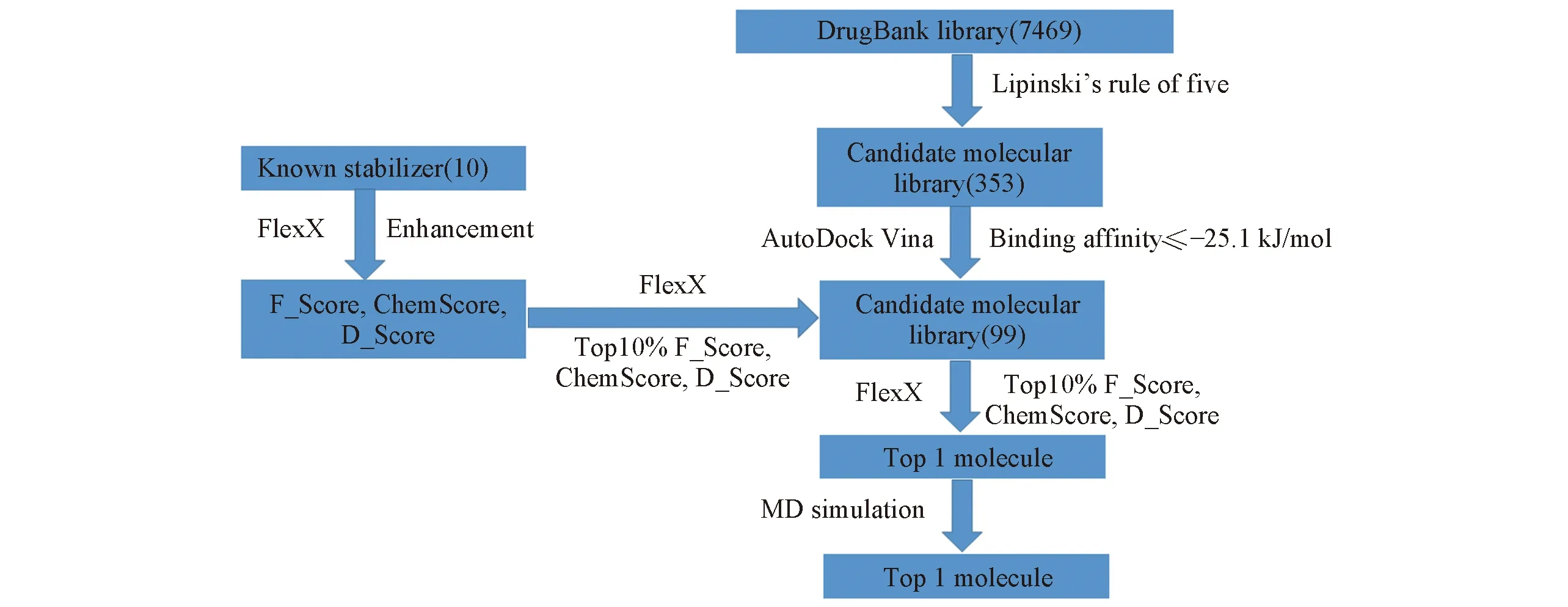
Fig.1 Procedure of virtual screening
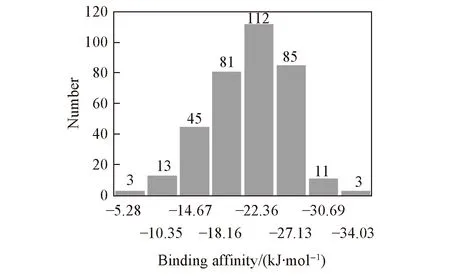
Fig.2 Binding affinity distribution of these small molecules and Y220C mutant of p53C analyzed by AutoDock Vina
从DrugBank 4.0数据库中选择7469个分子作为潜在的稳定剂, 考虑到p53C-Y220C表面的疏水腔很小, 因此稳定剂的分子量不能太大. Boeckler等[11]首先利用利宾斯基五原则筛选了Zinc数据库, 然后利用药效团模型和分子对接虚拟筛选获得了能够提高p53C-Y220C突变体的稳定剂PhiKan083. 因此, 我们首先利用DrugBank 4.0网站(http: //www.drugbank.ca/)[13]提供的利宾斯基五原则筛选获得了353个候选小分子(图1). 然后利用AutoDock Vina将这353个小分子分别与p53C-Y220C的空腔进行分子对接. 图2为353个小分子的亲和力(binding affinity)分布, 其中亲和力≤-25.1 kJ/mol的小分子共有99个. 而乙酰胆碱酯酶抑制剂他克林(Tacrine)(ID: DB00382)、 瑞莫必利(ID: DB00409)和2-Amino-6-Aminomethyl-8-Phenylsulfanylmethyl-3h-Quinazolin-4-One(ID: DB04239)与p53C-Y220C的亲和力最小(≤-33.5 kJ/mol).
现有的分子对接软件在构象搜索和对配基-目标蛋白质之间的亲和性评价时均存在优缺点[24,25], 从而造成仅利用单个分子对接软件进行虚拟筛选很容易引入较大的误差. 目前常用的解决办法是利用一种或多种分子对接软件进行虚拟筛选, 然后基于多种打分函数结果来综合评估配基与目标蛋白质之间的亲和作用力. 首先需要选取能够对已知稳定剂进行有效筛选的打分函数. 采用打分函数对已知稳定剂的富集率来考察这些打分函数, 并优先从数据库中筛选出已知稳定剂的性能[26,27]. 首先查找文献获得10个能够稳定结合到Y220C突变所导致的结合腔中的化合物(表S1, 见本文支持信息), 然后利用FlexX软件将这10个稳定剂和利用AutoDock Vina计算的结合自由能≤-25.1 kJ/mol的99个潜在的稳定剂分别与p53C-Y220C对接, 然后基于SYBYL所带的5种打分函数F_Score, ChemScore, D_Score, G_Score和PMF_Score来评价小分子和p53C-Y220C之间的亲和力.
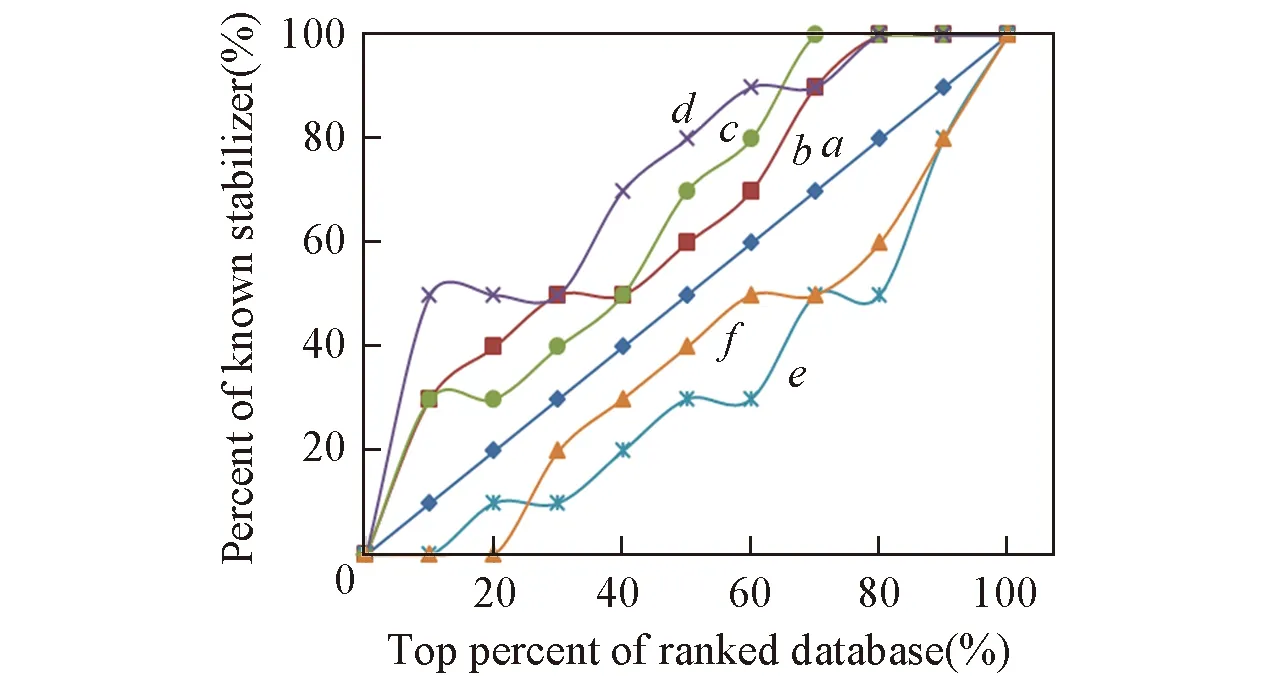
Fig.3 Enrichment curve analysis of the fivescoring functions in FlexXa. Random; b. F_Score; c. ChemScore; d. D_Score; e. G_Score; f. PMF_Score. A diagonal represents a random selection. Curves above the diagonal represent the corresponding scoring functions can select the known stabilizers from the library of small molecular compounds.
图3为已知10个稳定剂和99个潜在的稳定剂分子与p53C-Y220C对接结果的打分函数富集曲线. 从图3可以看出, 只有F_Score, ChemScore和D_Score 3种打分函数对已知的10种稳定剂有富集效果, 而另外2种打分函数G_Score和PMF_Score不能有效地从现有的数据库中将已知的稳定剂筛选出来. 因此, 第2次分子对接仅考虑F_Score, ChemScore和D_Score这3种打分函数的结果. 另外, 从图3可以清晰地看出, 当选用top 10%作为选择标准时, F_Score和ChemScore均能筛选出30%的稳定剂分子, 而使用D_Score更能获得高达50%的稳定剂分子. 因此, 本研究选择这3种打分函数的top 10%作为p53C-Y220C稳定剂的筛选标准. 即候选分子均排在这3种打分函数前10名之内才能作为p53C-Y220C的稳定剂. 表S2(见本文支持信息)列出利用3种打分函数计算得到的前10名的分子. 可以看出, 只有他克林(ID: DB00382)在这3种打分函数的排名中均进入前10. 因此, 他克林作为潜在的稳定剂用于后续研究. 而利用AutoDock Vina软件筛选中获得的另外2个亲和能力<8.0的分子瑞莫必利和2-Amino-6-Aminomethyl-8-Phenylsulfanylmethyl-3h-Quinazolin-4-One却不能满足上述筛选条件.
2.2 小分子与p53C突变体Y220C蛋白质复合物亲和性的分子动力学模拟验证
大量研究[28~32]表明, 仅利用分子对接筛选配基会存在大量的假阳性和假阴性结果. 这主要是由分子对接方法的缺陷所致: (1) 仅考虑配基的柔性, 而将目标蛋白质看作刚性; (2) 在计算配基-目标蛋白质之间的亲和作用过程中没有考虑溶剂化效应对配基-目标蛋白质之间亲和力的影响. 上述缺点决定了仅利用分子对接来筛选较高亲和性的分子会带来很大误差. 为了很好地弥补分子对接的上述缺点, 本文利用全原子MD模拟来进一步验证利用分子对接筛选获得的小分子和目标蛋白质之间的亲和力[25,33~35]. 由于利用AutoDock Vina软件虚拟筛选得到的瑞莫必利和2-Amino-6-Aminomethyl-8-Phenylsulfanylmethyl-3h-Quinazolin-4-One与目标蛋白质之间的亲和力远强于他克林, 但这2个分子却不能被F_Score, ChemScore和D_Score 3种打分函数所富集. 因此, 我们也利用MD模拟进一步验证了瑞莫必利和2-Amino-6-Aminomethyl-8-Phenylsulfanylmethyl-3h-Quinazolin-4-One与p53C-Y220C之间的亲和力.
图4(A)和(B)所示分别为分子对接(即MD模拟起始阶段的构象)和100 ns MD模拟后3个小分子和p53C-Y220C疏水腔的结合位置. 由图4(B)可见, 在100 ns MD模拟时间内, 只有他克林分子一直稳定结合在蛋白表面疏水空腔内. 通过比较100 ns MD模拟前后的构象可以看出, 他克林分子和p53-Y220C结合位置发生了改变. 他克林分子从疏水腔的表面移到疏水腔内部. 与之相反, 另外2个分子在100 ns MD模拟过程中均不能结合在这个疏水腔内. 瑞莫必利分子从3 ns后就从疏水腔中游离出来, 并在随后的模拟时间内没有结合到目标蛋白的任何部位[图4(B)]. 而DB04239分子从疏水空腔中慢慢移出而结合到了目标蛋白质的其它部位[图4(B)]. 模拟结果说明, DB04239分子与Y220C突变所形成的疏水腔之间的亲和性并不强, 而瑞莫必利分子则与疏水腔完全没有亲和性. 只有他克林与Y220C所形成的疏水空腔有较强的亲和作用. 进一步证明仅利用AutoDock Vina和该软件自带的打分函数进行亲和配基筛选具有很大的误差, 而基于多种打分函数结果的富集率进行虚拟筛选数据库可以较好的弥补上述缺点.
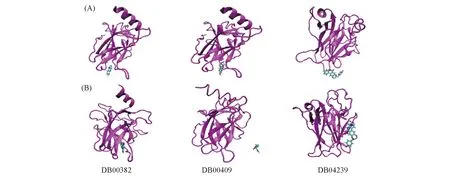
Fig.4 Comparison of binding sites of the 3 compounds on p53C mutant Y220C before and after MD simulations (A) Binding models obtained by docking(i.e., initial binding conformations in MD simulation); (B) binding models obtainedafter 100 ns MD simulations.
2.3 他克林与p53C-Y220C蛋白之间亲和作用力分析
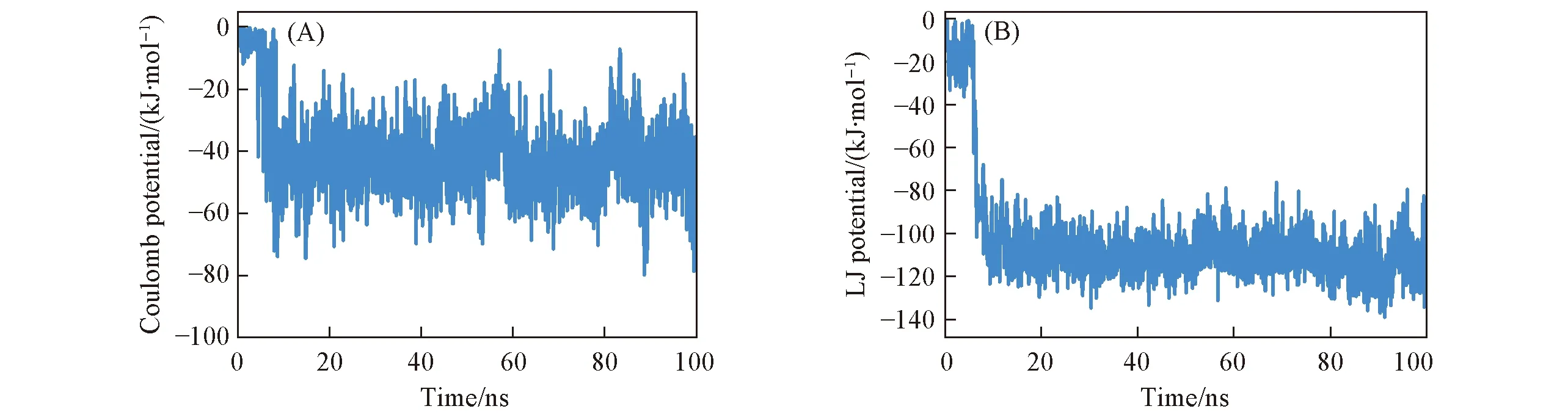
Fig.5 Time-dependent Coulomb(A) and LJ(B) potential energy of tacrine- p53C-Y200C
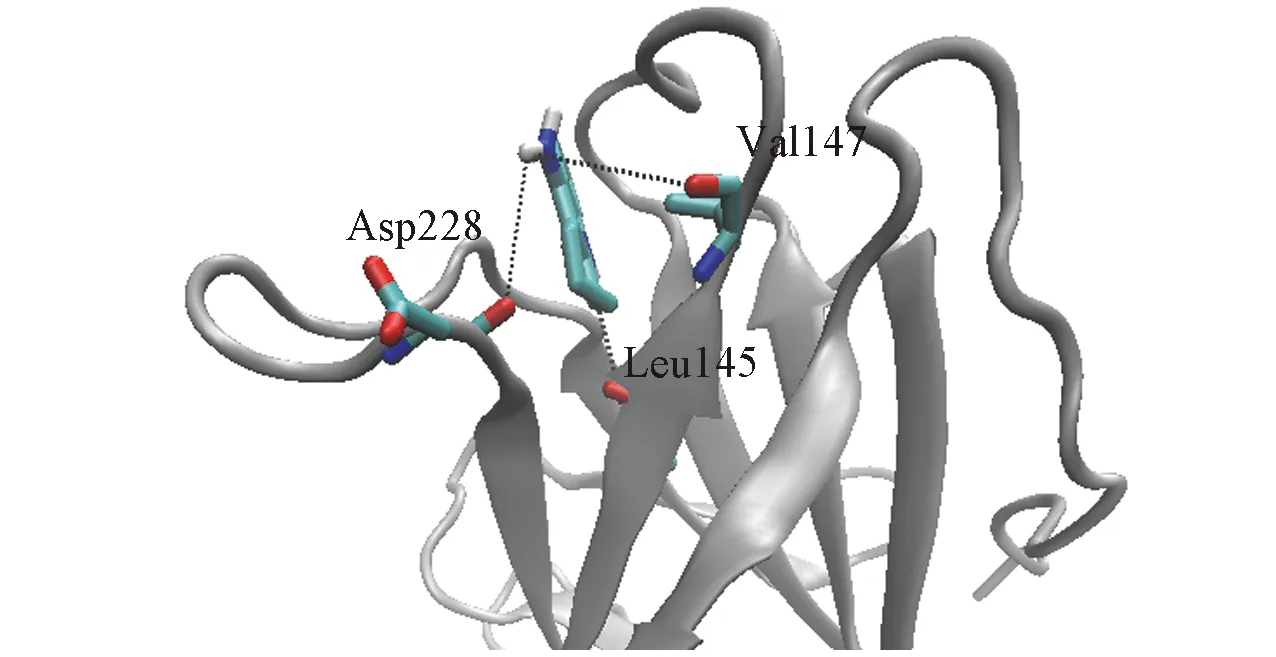
Fig.6 Snapshot of the hydrogen bonds betweentacrine and the residues of p53C-Y220CHydrogen bonds between tacrine and the residues Leu145, Val147 and Asp228 of p53C-Y220C are shown in black dotted lines. The residue type and its sequence number which interacted with tacrine via hydrogen bonds are also shown.
利用g_energy计算了他克林和p53C-Y220C之间的库仑(Coulomb)和LJ势能, 并示于图5. 其中, Coulomb和LJ势能分别代表他克林和目标蛋白质之间的静电和疏水相互作用. 势能的数值越低, 表示配基-目标蛋白质之间相应的作用力越强. 从图5可见, 他克林与目标蛋白之间Coulomb和LJ势能的变化趋势基本一致. 在初始的10 ns内, 他克林和目标蛋白质之间的Coulomb和LJ势能均发生急剧变化. 他克林和目标蛋白质之间的Coulomb势能从0急剧降低到-40 kJ/mol左右, 并在随后的90 ns内基本稳定在-50 kJ/mol. 虽然在初始的10 ns内, LJ势能的变化趋势和Coulomb势能类似, 但在最后90 ns内, 他克林和目标蛋白质之间的LJ势能降低到-110 kJ/mol. 即他克林和目标蛋白质之间的疏水作用力是其静电作用力的2倍. 因此, 他克林和p53C突变体Y220C之间的作用力主要为疏水和静电相互作用, 疏水相互作用占主导地位.
根据MD模拟获得的他克林和p53C-Y220C复合物的典型构象, 计算得出他克林和p53C-Y220C之间的氢键, 如图6所示. 从图6可见, 他克林与疏水空腔周围的3个氨基酸残基形成3个氢键, 从而使小分子稳定地结合在这个空腔内. 可以看出, 他克林的氨基基团作为氢供体, 它上面的氢原子可以分别与Leu147和Asp228肽键上的氧原子形成2个氢键. 同时, 他克林上的氮原子也可以接受Val145肽键上-NH基团的氢原子而形成另外1个氢键.
2.4 他克林与p53C突变体Y220C疏水腔的结合过程分析
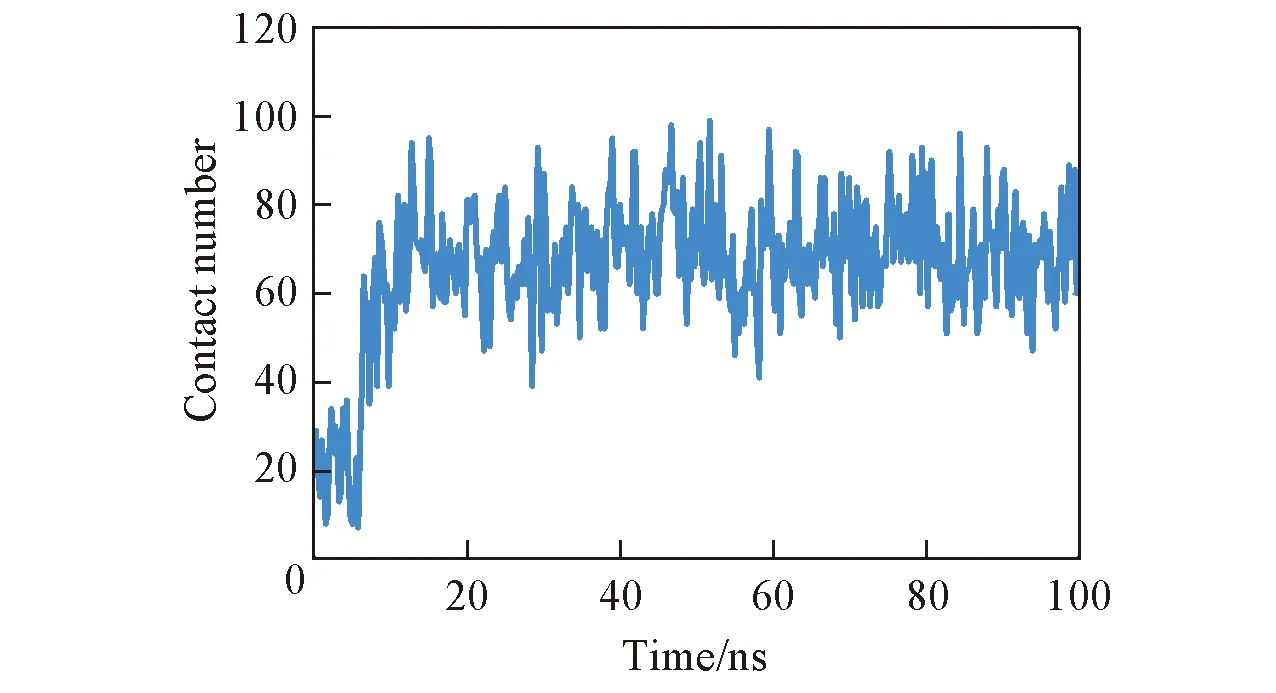
Fig.7 Time-dependent contact number between the tacrine and Y220C mutant of p53C
计算了他克林和目标蛋白质之间的接触数随模拟时间的变化(图7). 从图7可见, 在MD模拟的初始8 ns内, 他克林与p53C-Y220C之间接触数比较小(<40), 此时他克林仅有一部分与p53C-Y220C结合, 而另一部分暴露在疏水腔外(图8). 在初始的8 ns模拟时间内, 他克林与目标蛋白质的结合构象与分子对接所获得构象类似. 随着模拟的进行, 如图8中8 ns典型构象所示, S3/S4环与S7/S8环之间的狭缝发生结构微调, 从而使他克林分子逐渐向这个缝隙中移动. 与此同时,他克林和目标蛋白质之间的接触数急剧上升(图7). 为了与目标蛋白质发生亲和作用, 他克林在结合蛋白过程中的构象不断调整. 经过10 ns MD模拟, 他克林平行结合到Y220C的S3/S4环与S7/S8环所形成的缝隙中(图8). 在随后的MD模拟过程中, 他克林分子竖直插入到这个空腔, 整个小分子被Y220C突变所形成的空腔所包裹(图8). 此时, 蛋白质-小分子之间的接触数达到最大(图7). 在随后的90 ns内, 他克林均稳定地结合在目标蛋白Y220C突变所形成的空腔内, 接触数也相对稳定(图7). 这进一步说明他克林和p53C-Y220C之间具有非常强的亲和作用.
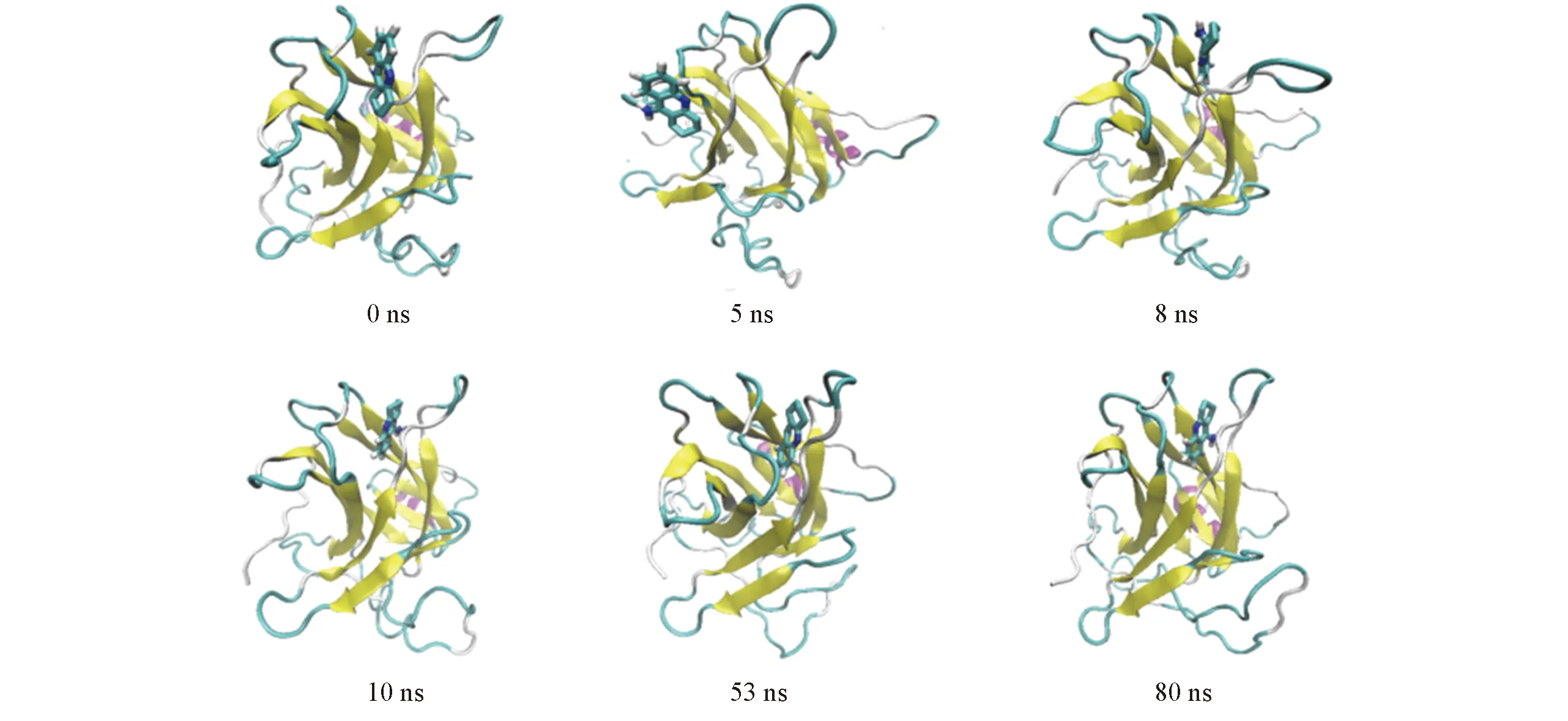
Fig.8 Representative conformations of tacrine and p53C-Y220C complex during the 100 ns MD simulations
图9所示为100 ns MD模拟后的他克林分子结合到p53C突变体Y220C疏水空腔中的示意图. 可以看出, 在他克林结合到p53C-Y220C突变所形成的疏水空腔后, 对L7/L8环的结构和位置造成很大的影响. 由于他克林主要通过疏水和静电作用力(包括氢键)与S3/S4环和L7/L8环上的氨基酸残基发生相互作用, 在他克林分子的介导作用下, S7/S8环逐渐靠近S3/S4环[图9(A)], 从而使p53C-Y220C突变体表面的疏水腔的容积逐渐减小, 由原先的打开状态变成了一个闭合的状态[图9(B)]. 从而使目标蛋白质表面Y220C的附近仅剩一个很小的空腔, 而他克林分子恰好位于突变产生的空腔中(图9). 因此, 他克林分子结合到这个空腔中, 提高了p53C-Y220C突变体的稳定性. 此外, 利用MD模拟研究了他克林和p53C-Y220C复合物在44 ℃的结构稳定性. 在折叠和去折叠过程中, 常用Cα原子的均方根偏差(RMSD)的数值来表示蛋白质结构与初始结构的变化[36,37]. 本研究也选择了Cα原子的RMSD来表示目标蛋白质的结构与初始结构之间的差别, 即RMSD值越大表示目标蛋白质的结构与初始结构差别越大. 图S3(见本文支持信息)列出了p53C-Y220C的Cα原子的RMSD值随模拟时间的变化. 可以看出, 结合他克林的p53C-Y220C的Cα的RMSD值平衡后基本维持在0.4 nm左右, 远低于不含他克林的目标蛋白质的Cα的RMSD值(0.6 nm). 由于目标蛋白质p53C-Y220C的结构稳定性较差, 在44 ℃下很快就发生构象转换, 从而使目标蛋白质的RMSD变化较大. 由于他克林对于目标蛋白质之间的稳定作用, 从而使目标蛋白质不容易发生构象转换, 因此该蛋白质的RMSD值变化较小. 这也进一步证明了他克林确实能够稳定p53C-Y220C的结构.
2.5 他克林提高p53C突变体Y220C稳定性的实验验证
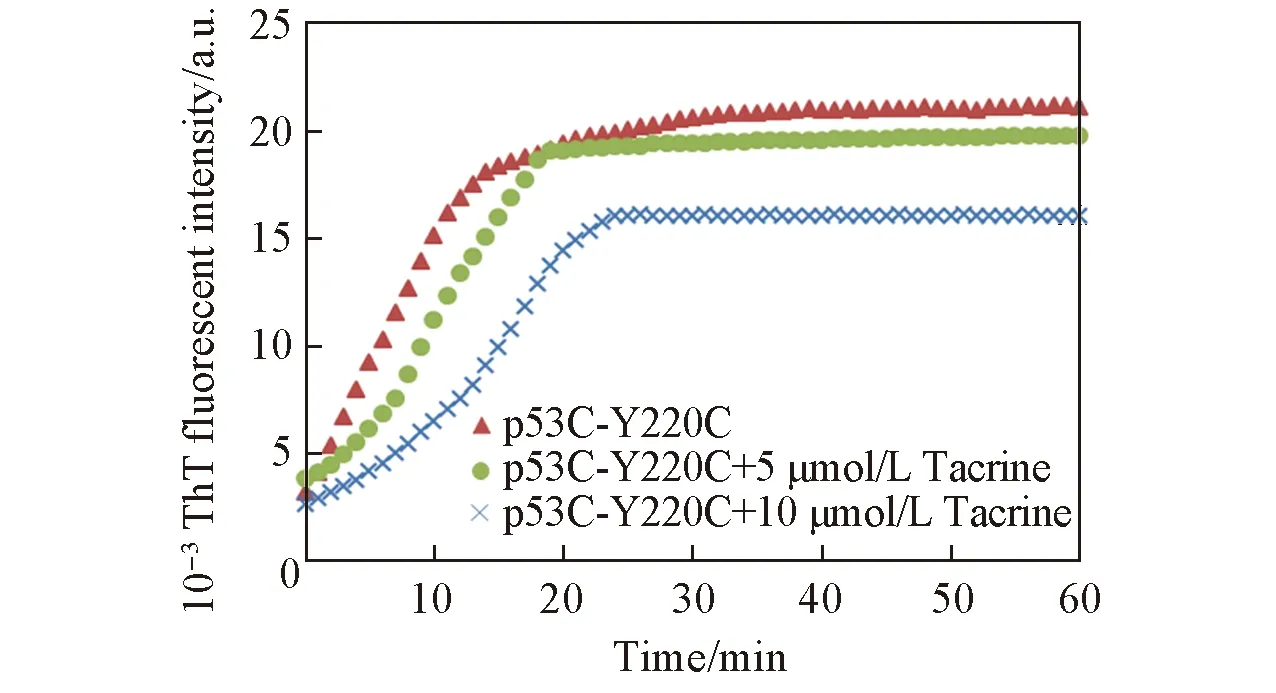
Fig.10 Effect of different concentrations of tarcrine on p53C-Y220C aggregation measured by ThT fluorescence The concentration of p53C-Y220C was 5 μmol/L.
研究表明, Y220C突变会进一步降低p53C的稳定性, 从而加速p53C-Y220C突变体聚集形成淀粉样纤维[7]. 由于这些淀粉样纤维中含有的大量β-折叠片能够特异地结合ThT而发荧光, 因此ThT荧光实验常被用于定量检测p53C-Y220C突变体的聚集程度[38]. 而由于p53C-Y220C的聚集沉淀是由其稳定性降低所引起的, 因此利用ThT荧光实验来验证他克林能否提高p53C突变体Y220C的稳定性. 首先利用大肠杆菌表达并依次通过阳离子交换色谱和肝素亲和色谱分离获得了p53C-Y220C蛋白质. 然后利用ThT荧光实验表征他克林抑制p53C-Y220C聚集的影响, 结果如图10所示.
当不含他克林分子时, ThT荧光强度在20 min内从初始的3000左右急剧增加到18600, 并经过缓慢增长期后到达平稳期, 其ThT荧光强度基本保持在21000左右. 在5 μmol/L他克林存在下, 在聚集初始的20 min内ThT荧光强度明显低于纯p53C-Y220C, 但最终的ThT荧光强度仅稍低于纯p53C-Y220C聚集体的ThT荧光强度. 即5 μmol/L他克林仅能够延缓p53C-Y220C的聚集, 并不能减少最终成熟纤维体的形成. 在10 μmol/L他克林存在下, 他克林表现出更强的抑制效果. 表明他克林不仅极大地延缓p53C-Y220C的聚集速度而且极大地减少了成熟纤维的形成量. 因此, 他克林能够提高p53C-Y220C的稳定性, 从而抑制p53C-Y220C的聚集, 且他克林提高p53C-Y220C稳定性的能力与其浓度密切相关. 这与常见的淀粉样蛋白质的聚集抑制剂是相同的[39,40].
3 结 论
基于利宾斯基五原则从DrugBank 4.0数据库中筛选获得了353个潜在的p53C-Y220C的稳定剂, 利用AutoDock Vina分子对接软件筛选获得了99个亲和力≤-25.1 kJ/mol的稳定剂分子. 利用FlexX软件结合5种打分函数筛选获得了p53C-Y220C的稳定剂他克林分子, 并利用全原子MD模拟进一步验证了他克林与p53C-Y220C之间的亲和性. 发现他克林分子能够稳定结合到Y220C所形成的疏水空腔中. 他克林和目标蛋白质之间的亲和作用力主要为疏水和静电相互作用, 而疏水相互作用占主导地位. 此外, 他克林还与p53C突变体Y220C疏水腔周围的3个残基Leu145, Val147和Asp228形成氢键作用. 在他克林和疏水腔周围残基之间的相互作用下, S7/S8环逐渐靠近S3/S4环, 从而使p53C-Y220C突变体表面的疏水腔的容积逐渐减小, 由原先的打开状态变成了一个闭合的状态, 从而大大提高了p53C突变体Y220C的稳定性. 利用ThT荧光实验证明他克林能够提高p53C-Y220C突变体的稳定性, 从而抑制了p53C-Y220C突变体的聚集.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20150790.
[1] Levine A. J., Oren M.,Nat.Rev.Cancer, 2009, 9(10), 749—758
[2] Olivier M., Hollstein M., Hainaut P.,CSHPerspect.Biol., 2010, 2(1), a001008
[3] Xu Z. Y., Zhao L. L., Cao Z. X., Wang J. H.,ActaPhys-Chim.Sinica, 2012, 28(7), 1665—1675(许朝莹, 赵立岭, 曹赞霞, 王吉华. 物理化学学报, 2012, 28(7), 1665—1675)
[4] Cho Y., Gorina S., Jeffrey P. D., Pavletich N. P.,Science, 1994, 265(5170), 346—355
[5] Joerger A. C., Fersht A. R.,Annu.Rev.Biochem., 2008, 77, 557—582
[6] Joerger A. C., Ang H. C., Fersht A. R.,P.Natl.Acad.Sci.USA, 2006, 103(41), 15056—15061
[7] Wang G., Fersht A. R.,P.Natl.Acad.Sci.USA, 2015, 112(8), 2437—2442
[8] Wilcken R., Liu X., Zimmermann M. O., Rutherford T. J., Fersht A. R., Joerger A. C., Boeckler F. M.,J.Am.Chem.Soc., 2012, 134(15), 6810—6818
[9] Bykov V. J., Wiman K. G.,FEBSLett., 2014, 588(16), 2622—2627
[10] Friedler A., Hansson L. O., Veprintsev D. B., Freund S. M., Rippin T. M., Nikolova P. V., Proctor M. R., Rudiger S., Fersht A. R.,P.Natl.Acad.Sci.USA, 2002, 99(2), 937—942
[11] Boeckler F. M., Joerger A. C., Jaggi G., Rutherford T. J., Veprintsev D. B., Fersht A. R.,P.Natl.Acad.Sci.USA, 2008, 105(30), 10360—10365
[12] Wassman C. D., Baronio R., Demir O., Wallentine B. D., Chen C. K., Hall L. V., Salehi F., Lin D. W., Chung B. P., Hatfield G. W., Richard Chamberlin A., Luecke H., Lathrop R. H., Kaiser P., Amaro R. E.,Nat.Commun., 2013, 4, 1407
[13] Law V., Knox C., Djoumbou Y., Jewison T., Guo A. C., Liu Y. F., Maciejewski A., Arndt D., Wilson M., Neveu V., Tang A., Gabriel G., Ly C., Adamjee S., Dame Z. T., Han B. S., Zhou Y., Wishart D. S.,NucleicAcidsRes., 2014, 42(D1), D1091—D1097
[14] Trott O., Olson A. J.,J.Comput.Chem., 2010, 31(2), 455—461
[15] Rarey M., Kramer B., Lengauer T., Klebe G.,J.Mol.Biol., 1996, 261(3), 470—489
[16] Schuttelkopf A. W., van Aalten D. M.,ActaCrystallogr.D.Biol.Crystallogr., 2004, 60(8), 1355—1363
[17] Oostenbrink C., Villa A., Mark A. E., van Gunsteren W. F.,J.Comput.Chem., 2004, 25(13), 1656—1676
[18] Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E.,Bioinformatics, 2013, 29(7), 845—854
[19] Darden T., York D., Pedersen L.,J.Chem.Phys., 1993, 98, 10089—10092
[20] Hess B., Bekker H., Berendsen H. J. C.,J.Comput.Chem., 1997, 18, 1463—1472
[21] Beredsen H. J. C., Postma J. P. M., van Gunsteren W. F., Di Nola A., Haak J. R.,J.Chem.Phys., 1984, 81, 3684—3690
[22] Bussi G., Donadio D., Parrinello M.,J.Chem.Phys., 2007, 126(1), 014101
[23] Humphrey W., Dalke A., Schulten K.,J.Mol.Graph.Model., 1996, 14(1), 33—38
[24] Liu F. F., Wang T., Dong X. Y., Sun Y.,J.Chromatogr.A, 2007, 1146(1), 41—50
[25] Zhao W. W., Liu F. F., Shi Q. H., Dong X. Y., Sun Y.,Biochem.Eng.J., 2014, 88, 1—11
[26] Marsh L.,PlosOne, 2011, 6(8), e23215
[27] Du W. J., Guo J. J., Gao M. T., Hu S. Q., Dong X. Y., Han Y. F., Liu F. F., Jiang S., Sun Y.,Sci.Rep., 2015, 5, 7992
[28] Chen Y. C.,TrendsPharmacol.Sci., 2015, 36(2), 78—95
[29] Hou T. J., Wang J. M., Li Y. Y., Wang W.,J.Comput.Chem., 2011, 32(5), 866—877
[30] Wu Y. J., Cui Y. L., Zheng Q. C., Zhang H. X.,Chem.J.ChineseUniversities, 2014, 35(12), 2605—2611(吴云剑, 崔颖璐, 郑清川, 张红星. 高等学校化学学报, 2014, 35(12), 2605—2611)
[31] Dong L., Yi Z. S., Wu Z. W., Wang H. Y., Zhang A. Q.,Chem.J.ChineseUniversities, 2015, 36(3), 516—522(董露, 易忠胜, 伍智蔚, 王海洋, 张爱茜. 高等学校化学学报, 2015, 36(3), 516—522)
[32] Jian W. J., Zeng Y., Xiong H. J., Pang J.,Carbohydr.Polym., 2011, 85 452—456
[33] Bai S., Zhou R., Liu F. F.,ActaPhys-Chim.Sinica, 2013, 29(2), 439—448(白姝, 周荣, 刘夫锋. 物理化学学报, 2013, 29(2), 439—448)
[34] Lin D. Q., Tong H. F., Wang H. Y., Yao S. J.,J.Phys.Chem.B, 2012, 116(4), 1393—1400
[35] Wu F., Yang Z. W., Yuan X. H.,Chem.J.ChineseUniversities, 2013, 34(4), 931—938(武菲, 杨志伟, 袁晓辉. 高等学校化学学报, 2013, 34(4), 931—938)
[36] Zhang N., Liu F. F., Dong X. Y., Sun Y.,J.Phys.Chem.B, 2012, 116(24), 7040—7047
[37] Liu F. F., Dong X. Y., Sun Y.,J.Mol.Graph.Model., 2008, 27(4), 421—429
[38] Wang G., Fersht A. R.,P.Natl.Acad.Sci.USA, 2015, 112(8), 2443—2448
[39] Liu F. F., Dong X. Y., Sun Y.,ActaPhys-Chim.Sinica, 2010, 26(6), 1643—1650(刘夫锋, 董晓燕, 孙彦. 物理化学学报, 2010, 26(6), 1643—1650)
[40] Xiong N., Dong X. Y., Zheng J., Liu F. F., Sun Y.,ACSAppl.Mater.Inter., 2015, 7(10), 5650—5662
(Ed.: Y, Z)
† Supported by the National Natural Science Foundation of China(No.21576199) and the China Postdoctoral Science Foundation(Nos.2012T50241, 2013M530115).
Virtual Screening of Small Molecular Stabilizer for Y220C Mutant of p53†
DING Jiyong, SHEN Hongchen, LIU Fufeng*
(KeyLaboratoryofSystemsBioengineeringoftheMinistryofEducation,DepartmentofBiochemicalEngineering,SchoolofChemicalEngineeringandTechnology,TianjinUniversity,Tianjin300072,China)
About half of cancers are caused by the mutation in the tumor suppressor p53. A hydrophobic cavity on the surface of p53 was caused by the Y220C mutation. The surface crevice is on the opposite side and distant from the DNA-binding domain, making it a particularly attractive target site for stabilizing small-molecule drugs. In order to obtain the effective stabilizers, Lipinski’s rule of five, twice docking methods and molecular dynamics(MD) simulations were successively used for virtual screening the DrugBank 4.0 library and tacrine was obtained as a candidate stabilizer. Then, all-atom MD simulations were used to verify the affinity between tacrine and the target protein. The MD simulation indicated that tacrine bound tightly to the pocket and the complex remained stable. The affinity between tacrine and the target protein was further analyzed. It was found that the hydrophobic and electrostatic interactions dominated the affinity between tacrine and the target protein. And, the hydrophobic interactions were dominant force. Moreover, there were 3 hydrogen bonds between tacrine and the residues Leu145, Val147 and Asp228 of p53C-Y220C. And then, the detailed binding process of tacrine and p53C-Y220C was probed based on MD simulations. Finally, the stabilizing capacity of tacrine on p53C-Y220C was further validated by thioflavine T fluorescent experiments.
Cancer; Protein stabilizer; Molecular dock; Molecular dynamics simulation; p53; Tacrine
10.7503/cjcu20150790
2015-10-13.
日期: 2016-03-18.
国家自然科学基金(批准号: 21576199)和中国博士后科学基金(批准号: 2013M530115, 2012T50241)资助.
O641
A
联系人简介: 刘夫锋, 男, 博士, 副教授, 从事生物过程的分子模拟研究. E-mail: fufengliu@tju.edu.cn
猜你喜欢
儿童故事画报·发现号趣味百科(2024年4期)2024-05-18 13:04:39
石油沥青(2021年4期)2021-10-14 08:50:54
装备制造技术(2021年2期)2021-07-21 05:38:08
水利科技与经济(2017年5期)2017-04-22 02:39:36
发明与创新·中学生(2017年1期)2017-01-20 20:37:52
发明与创新(2017年2期)2017-01-18 08:48:21
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:26
中国塑料(2015年3期)2015-11-27 03:42:18
中国塑料(2015年3期)2015-11-27 03:42:16
中国塑料(2015年10期)2015-10-14 01:13:22
