
遗传关联性研究及其Meta分析的简介
2016-11-23 09:08翁鸿李妙竹耿培亮曾宪涛
中国循证心血管医学杂志 2016年10期
翁鸿,李妙竹,耿培亮,曾宪涛
遗传关联性研究及其Meta分析的简介
翁鸿1,2,李妙竹3,耿培亮1,曾宪涛1,2
基因因素在疾病的发生发展中扮演着重要的角色,遗传关联性研究是基因与人类复杂疾病的重要研究方法之一。但在关联分析领域存在一种“赢者诅咒”现象,此外,由于单个基因效应对大多数疾病来说均较小,因此,需要大样本研究来探索其真实效应。本文主要简要介绍遗传关联性研究及其Meta分析的方法以及报告规范。
候选基因;遗传关联性研究;基因多态性;Meta分析
现代遗传学研究的核心是通过研究何种遗传变异导致疾病表型,从而阐明发病机制,并由此机制出发研究可能干预或预防疾病的方法[1]。虽然目前我们距离这一目标还较遥远,但也取得了显著的进步。复杂疾病的经典研究方法主要是连锁分析和关联分析。近几十年来,随着高通量基因型检测技术的发展,越来越多的有关疾病遗传易感性的关联分析研究发表。但大多数起始阳性结果的研究并不具有可重复性[2,3],特别是候选基因法的研究,即“赢者诅咒”现象[4]。这可能是由于这些原始研究的结果为假阳性结果,也可能是大多数后续研究的统计效能不足以检测出较小的基因效应。此外,研究间异质性也是影响结果的重要因素。有研究显示,55篇候选基因研究Meta分析的平均比值比为1.33,因此,遗传关联研究需要成千上万的样本量来检测较小的基因效应[5]。而在实际研究中,大样本量的收集较难实现,且费用高。
而Meta分析是一种合并同一研究主题的单个研究的统计学方法,具有更高的统计学效能,并能增加结果的可信度和精确性[6,7]。此外,Meta分析还可以检测研究间异质性,检测和量化研究间异质性是基因-疾病Meta分析研究的重要部分,处理研究间异质性可能会给研究结果带来新的视角[8,9]。本文主要介绍遗传关联性研究Meta分析的一些相关问题,供研究者参考。
1 遗传关联性研究简介
遗传关联性研究主要有两大目的[10]:①基因识别分析;②基因特征分析。前者多数是由遗传学家开展的,研究设计主要是基于人群或基于家系,主要解决的问题是某疾病是否有遗传成分。后者主要量化已识别出的基因变异或多态性与疾病的关联大小,多数是由流行病学家开展的基于人群的研究。
遗传关联性研究通过分析遗传变异和疾病的相关性,鉴别所选取的候选基因或染色体区域是否参与疾病的发生。如果某种疾病的个体中某变异基因的等位基因或基因型出现频率较高,我们认为该变异基因增加了该疾病的发病风险,即易感基因。其中单核苷酸多态性(SNP)是遗传关联研究使用最为广泛的检测指标,也是数量最多的,其他的遗传标记有微卫星、连续重复片段、插入/缺失及拷贝数变异。
2 遗传关联性研究的主要研究方法
遗传关联性研究主要有两种方法:一是候选基因法(candidate gene approach),二是全基因组关联研究(GWAS)。候选基因法的研究设计类型为基于人群或家系研究,多数采用病例-对照研究进行。候选基因法是建立在目前对某疾病的生物学和病理生理学的理解之上进行的,即某基因的产物参与某疾病的发生过程,从而检测其变异是否与该疾病具有相关性。该方法是近十几年来检测遗传变异和疾病相关性的主要方法,特别是在基因-环境交互作用的背景下,该方法是主要的方法。其主要的缺点在于初始阳性研究的结果的可重复性较差,即初始发表的研究可能会趋向于高估效应量,而后续研究对该变异与疾病易感性的评估趋于缓和,这被称为“赢者诅咒”现象[4]。
后基因组时代的到来,使得GWAS得以实现,当前已有较多的GWAS研究发表,如具有代表性的老年性黄斑变性[11,12]、Ⅰ型糖尿病[13,14]和开角型青光眼[15]。GWAS是一种假设-生成的方法[1]。通过全基因组扫描,确定新的与表型(疾病或性状)相关的遗传变异,不需要建立在任何生物学的基础之上,有时甚至是意外发现的某基因与某表型相关。GWAS的价值在于识别与疾病明确相关的新的遗传变异,而不是来量化这一关联效应。这也是其与候选基因法的区别之一。因此,GWAS和候选基因法可以相互补充,共同揭开复杂疾病的面纱。
3 遗传关联性研究Meta分析的分类
Sagoo等[16]将遗传关联性研究Meta分析按照数据来源主要分为基于文献和基于协作的Meta分析两大类(表1),其中基于文献的Meta分析分为未联系作者的Meta分析和联系作者的Meta分析,基于协作的Meta分析分为对现有数据的协作Meta分析和前瞻性数据的协作Meta分析。这主要的两种Meta分析各有利弊,可以相互补充。
此外,按照遗传关联性研究的研究方法可以分为候选基因研究的Meta分析和GWAS的Meta分析。候选基因研究的Meta分析最为常见。以“meta-analysis”和“polymorphism”为自由词检索PubMed,检索时限为2016年2月16日,初步检索结果为9070篇,其发表趋势如图1所示。
以“meta-analysis”为自由词结合“genomewide association study”检索PubMed,“genomewide association study”限制在“title/abstract”,检索时限为2016年2月16日,初步检索结果为745篇,其发表趋势如图2所示。当然,这二者的数据可能会有重叠的部分,也难免会有遗漏的部分。GWAS的Meta分析较少,这与GWAS技术成熟较晚有关,且GWAS的Meta分析方法也不完善,发展较慢。
4 遗传关联性研究及其Meta分析的报告规范
遗传关联性研究的报告规范主要有STREGA声明[17]。STREGA声明是Little等于2009年在STROBE声明的基础上提出的,旨在加强遗传关联性研究报告的透明化,其开发源自跨学科小组的商讨,多种学科的专家以及研究生参与。
目前尚未有统一正式的遗传关联性研究Meta分析报告规范,可以参考PRISMA声明[18]或MOOSE声明[19]。此外,人类基因组流行病学网络(HuGENet)发展了一种人类基因组流行病学评价(HuGE Review)的格式,属系统评价范畴,并制作了相关的手册(The HuGENetTM HuGE Review Handbook),其网址链接如下:http://www.med.uottawa.ca/public-health-genomics/ web/assets/documents/HuGE_Review_Handbook_ V1_0.pdf#x2122; Handbook of Systematic Reviews [PDF 166KB]。目前要求研究者按照HuGE Review格式撰写论文的期刊有American Journal of Epidemiology、American Journal of Obstetrics and Gynecology、Archives of Dermatology、Cancer Epidemiology Biomarkers & Prevention、Gastroenterology、Genetics in Medicine、International Journal of Epidemiology、Paediatric and Perinatal Epidemiology。此外,2014年PLoS One杂志的编辑在社区微博上发表了针对遗传关联性研究Meta分析的清单[20],要求投稿作者填写此清单并回答相关问题。同年,American Journal of Epidemiology杂志声明支持PLoS One杂志的这一清单,并更新了HuGE Review的部分内容,要求投稿作者按照此标准进行撰写遗传关联性研究的Meta分析[21]。
5 小结
遗传关联性研究是众多复杂疾病研究设计的基础,其重要性不言而喻。其缺点刚好可以运用Meta分析来完善。但近几年的遗传关联性研究Meta分析多为人诟病,原因在于其数量呈指数增长,且质量较差,但不能因为质量差的遗传关联性研究的Meta分析而忽略其本身的作用,这是一把双刃剑,制作及使用Meta分析的人才是最关键的,而此时PLoS ONE、American Journal of Epidemiology杂志及HuGENet工作组均对该类型的Meta分析提出了撰写指导及要求,笔者呼吁研究者们制作遗传关联性研究Meta分析时能参考HuGE review手册,甚至是制作HuGE Review,这是值得推崇的。此外,在GWAS蓬勃发展的今天,也有人质疑候选基因法是否还能为遗传关联性研究做贡献,这点我们在第二部分已做相关阐述,候选基因法与GWAS各有优劣,相互补充,共同促进遗传学的发展、揭开复杂疾病的面纱。

表1 遗传关联性研究Meta分析的分类(按数据来源)
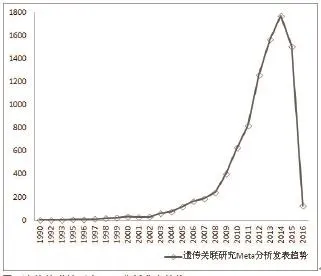
图1 遗传关联性研究Meta分析发表趋势
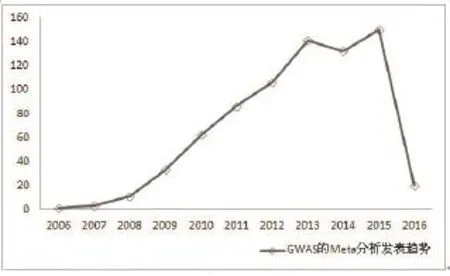
图2 GWAS的Meta分析发表趋势
[1] Al-Chalabi A,Almasy L. Genetics of Complex Human Diseases: A Laboratory Manual[M]. New York: Cold Spring Harbor Laboratory Press,2009.
[2] Ioannidis JP,Ntzani EE,Trikalinos TA,et al. Replication validity of genetic association studies[J]. Nat Genet,2001,29(3):306-9.
[3] Colhoun HM,McKeigue PM,Davey SG. Problems of reporting genetic associations with complex outcomes[J]. Lancet,2003,361(9360):865-72.
[4] Nakaoka H,Inoue I. Meta-analysis of genetic association studies: methodologies, between-study heterogeneity and winner's curse[J]. J Hum Genet,2009,54(11):615-23.
[5] Ioannidis JP. Genetic associations: false or true?[J]. Trends Mol Med,2003,9(4):135-8.
[6] 曾宪涛,冷卫东,郭毅,等. Meta分析系列之一:Meta分析的类型[J].中国循证心血管医学杂志,2012,4(1):3-5.
[7] 曾宪涛,邝心颖,孙燕,等. 什么是循证医学?[J]. 湖北医药学院学报,2013,32(1):1-5.
[8] Lau J,Ioannidis JP,Schmid CH. Summing up evidence: one answer is not always enough.[J]. Lancet,1998,351(9096):123-7.
[9] Ioannidis JP,Patsopoulos NA,Evangelou E. Heterogeneity in metaanalyses of genome-wide association investigations[J]. PLoS One,2007,2(9):e841.
[10] Thakkinstian A,McElduff P,D'Este C,et al. A method for meta-analysis of molecular association studies[J]. Stat Med,2005,24(9):1291-306.
[11] Edwards AO,Ritter RR,Abel KJ,et al. Complement factor H polymorphism and age-related macular degeneration[J]. Science, 2005,308(5720):421-4.
[12] Klein RJ,Zeiss C,Chew EY,et al. Complement factor H polymorphism in age-related macular degeneration[J]. Science,2005,308(5720):385-9.
[13] Cooper JD,Walker NM,Healy BC,et al. Analysis of 55 autoimmune disease and type II diabetes loci: further confirmation of chromosomes 4q27, 12q13.2 and 12q24.13 as type I diabetes loci, and support for a new locus, 12q13.3-q14.1[J]. Genes Immun,2009,10(Suppl 1):S95-120.
[14] Cooper JD,Walker NM,Smyth DJ,et al. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genomewide association study in type I diabetes families[J]. Genes Immun,2009,10(Suppl 1):S85-94.
[15] Bailey JN,Loomis SJ,Kang JH,et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma[J]. Nat Genet,2016,48(2):189-94.
[16] Sagoo GS,Little J,Higgins JP. Systematic reviews of genetic association studies. Human Genome Epidemiology Network[J]. PLoS Med,2009,6(3):e28.
[17] Little J,Higgins JP,Ioannidis JP,et al. Strengthening the reporting of genetic association studies (STREGA): an extension of the strengthening the reporting of observational studies in epidemiology (STROBE) statement[J]. J Clin Epidemiol,2009,62(6):597-608.
[18] Moher D,Liberati A,Tetzlaff J,et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement[J]. PLoS Med,2009,6(7):e1000097.
[19] Stroup DF,Berlin JA,Morton SC,et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group[J]. JAMA, 2000,283(15):2008-12.
[20] Editors (editors). Meta-analyses of genetic association studies-PLOS ONE's approach.http://blogs.PLOS.org/everyone/2014/04/04/ metaanalyses-genetic-association-studies-PLOS-ones-approach/.
[21] Gwinn M,Ioannidis JP,Little J,et al. Editorial: Updated guidance on human genome epidemiology (HuGE) reviews and meta-analyses of genetic associations[J]. Am J Epidemiol,2014,180(6):559-61.
本文编辑:姚雪莉
Brief introduction of genetic association studies and corresponding Meta-analysis
WENG Hong*, LI Miaozhu, GENG Pei-liang, ZENG Xian-tao.
*Center for Evidence-Based and Translational Medicine, Zhongnan Hospital, Wuhan University, Wuhan 430071, China.
ZENG Xian-tao, E-mail: zengxiantao1128@163.com
Gene plays an important role in the development and progression of diseases. The genetic association study is one of the vital approaches in the field of association between gene and complex diseases in human. However, a “winner’s curse” phenomenon is existed in the field of association study. In addition, the effect of each gene is relatively mild, therefore, large size sample studies are warranted to detect the true gene effect. The aim of this paper is to give a brief introduction of methods and reporting guideline of the genetic association studies and corresponding Meta-analyses.
Candidate gene; Genetic association study; Gene polymorphism; Meta-analysis
R4
A
1674-4055(2016)10-1156-03
国家重点研发计划专项基金(2016YFC0106300)
1430071 武汉,武汉大学中南医院循证与转化医学中心;2430071 武汉,武汉大学循证与转化医学中心;3北卡罗来纳州,美国杜克大学人群健康衰老研究中心
曾宪涛,E-mail:zengxiantao1128@163.com
10.3969/j.issn.1674-4055.2016.10.02
猜你喜欢
区域治理(2022年40期)2022-11-27
小学教学参考(语文)(2022年3期)2022-05-26
疯狂英语·新读写(2021年10期)2021-12-07
云南医药(2020年5期)2020-10-27
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
奥秘(2019年8期)2019-08-28
中国医药指南(2017年3期)2017-11-13
商周刊(2017年7期)2017-08-22
