
Mo(001)表面的CaO(001)薄膜模型催化体系的STM研究
2016-11-18 07:28利施宏刘慧慧邵
物理化学学报 2016年1期
王 利施 宏刘慧慧邵 翔,*吴 凯
(1中国科学技术大学化学物理系,合肥 230026;2北京大学化学与分子工程学院,北京 100871)
Mo(001)表面的CaO(001)薄膜模型催化体系的STM研究
王 利1施 宏1刘慧慧1邵 翔1,*吴 凯2,*
(1中国科学技术大学化学物理系,合肥 230026;2北京大学化学与分子工程学院,北京 100871)
氧化物单晶化薄膜的制备与表征是研究氧化物表面性质的重要方法,也是模型催化研究的前沿领域。本文主要综述了Fritz-Haber研究所的Hajo Freund小组在过去几年间围绕着以Mo(001)为衬底制备的CaO(001)薄膜模型催化体系而进行的表面结构和化学性质的系列研究。其中既包含了氧化物薄膜研究的共同特点,如界面效应、膜厚效应等,也包含有CaO/Mo体系独特的性质,如Mo的自发掺杂对表面性质的调控作用。在该系列研究中低温扫描隧道显微镜(LT-STM)技术的应用贯穿了方方面面,从原子结构表征到电子性质研究,从杂质缺陷的鉴别到表面物种荷电性质的分析等。STM所获得的微观信息直接从原子分子水平揭示了调控薄膜表面性质的各种控因。特别的,在理论计算的辅助下,不断深化认识氧化物掺杂调控的原理和机制,为设计新型催化剂提供重要思路。
CaO;薄膜;模型催化;STM;表面化学
1 引 言
碱土金属在地球上有着非常广泛的分布,由于化学活性高,通常均以化合物形式存在。碱土金属氧化物中除了BeO以六方纤锌矿结晶外,其他碱土金属氧化物均以离子型的石盐晶体形式进行结晶,通常都具有较高的熔点并在耐热材料中应用广泛。这种晶型中阴阳离子均按照面心立方结构排布并嵌套在一起,结构简单,因此非常适合作为模型体系进行研究(见图1)。MgO可能是模型催化中研究的最深入透彻的氧化物1,尽管其催化性质在一系列氧化物中是最差的,但是因为其结构简单,稳定性高,故而有利于实验和理论的研究。体相MgO是具有约7.8 eV带隙的绝缘体材料,但是生长在金属衬底上的超薄膜体系往往表现出足够的导电性,从而使得各种传统的电子谱学表征手段得以发挥应用。近年来人们在Ag(001)、Mo(001)、Fe(0001)、Au(111)等衬底上均实现了MgO(001)薄膜的制备,使各种表面科学及模型催化的相关研究成为可能,从实验和理论上对该薄膜体系进行了系统全面的研究。在其中相当一部分实验研究中,扫描隧道显微镜(STM)技术特别是低温STM(LT-STM)发挥了非常重要的作用,不但能够直接表征MgO表界面的结构信息,也能有效地获得具有空间分辨的电子结构信息,从而深化了人们对于MgO表面的构效关系的认识2。但是需要同时指出的是STM技术也存在化学分辨和时间分辨能力弱,对样品表面导电性和平整度要求高等方面的局限性,因此往往需要同其他谱学技术特别是理论计算结合起来才能更有效地揭示表面的物理化学性质。另外值得一提的是,氧化物薄膜技术的快速发展使得各种类型的氧化物材料均得以获得研究,特别的,薄膜体系具有特殊的结构和表面性质使其自身也形成了一种新型材料,展现出广阔的应用前景3。

图1 碱土金属氧化物所共有的食盐型离子晶体结构Fig.1 Rock-salt crystal structure of alkaline-earth metal oxide
在系列碱土金属氧化物中,CaO应该是最接近人们生活的。建筑中常用到的生石灰是制作水泥时的关键材料之一,其化学成分即CaO(而人们用的石灰水则是Ca(OH)2)。在各种玻璃中,CaO被广泛用作调节剂使用。CaO与MgO的结构及物理性质都很接近,然而其化学反应性远强于后者,因而在催化上也得到更加广泛的应用。如CaO被广泛应用于CO2、NO、SO2等酸性气体的吸附,以及作为甲烷氧化偶联反应、NO2还原反应、卤代烷烃的分解反应等反应的催化剂4–7。另外,作为对人体无害的金属氧化物,CaO在生物柴油的制备中也被广泛用作碱性催化剂8–12。尽管有着诸多的应用背景,关于CaO的表面科学研究却严重不足,少数基于单晶表面的研究直到最近才出现。如Brown等13,14及Kadossov和Burghaus15,16分别基于同步辐射技术对CaO(001)单晶表面及其对CO2、H2O等的吸附行为进行了初步探索。这种局面一方面是由于氧化物表面科学研究的整体滞后的现状所造成,另一方面也与CaO材料本身是绝缘体(带隙约7.1 eV)难以应用各种常规的电子谱学手段进行有效研究有关。在这样的背景之下,并且根据与MgO薄膜体系进行对比研究的需要,Fritz-Haber研究所的Hajo Freund小组自2010年开始着手利用包括STM、俄歇电子能谱(AES)、X射线光电子能谱(XPS)、反射吸收红外光谱(RAIRS)等谱学手段结合精细的理论计算,探索和研究CaO薄膜的制备及其表面与分子及金属的相互作用等。本文将主要概述该小组在上述方向上基于LT-STM为主要表征手段的相关研究进展。
2 CaO薄膜的生长制备
最早关于CaO薄膜的STM研究是Norenberg和Harding17在TiO2(110)上发现单晶中的杂质Ca元素在表面富集形成CaO超薄膜。然而这种通过表面偏析所形成的CaO薄膜其结构与性质均与块体材料差别较大,难以控制制备和深入研究。自上世纪末起,Maus-Friedrichs等18,19通过将金属Ca薄膜氧化的方法在Si(100)表面成功制备了CaO薄膜,并利用包括紫外光电子能谱(UPS),中等能量离子散射谱(MEIS),XPS等在内的各种技术研究了H2O、O2、CO2等小分子在其表面的吸附行为。但是由于缺乏STM技术的有效运用,这种薄膜的表面结构并不是很清晰,而且原子分子水平的微观信息也极度匮乏。其余的关于CaO薄膜的尝试研究参见GaN表面的多晶化薄膜20和Mo(001)表面的MgCaxO1–x混合型薄膜等的研究21。在这些研究中CaO薄膜无论就表面结构还是表面化学性质都没有受到很好的控制和研究。Hajo Freund小组具有长期研究MgO(001)薄膜的经验,成功地在Ag(001)和Mo(001)等单晶表面制备了MgO(001)薄膜。考虑到CaO与MgO在结构上的相似性,因此首先选择了Mo(001)作为衬底,并采用类似MgO薄膜的制备方法22,即在氧气气氛中将金属Ca蒸镀到Mo衬底表面,随后在超高真空环境中进行退火处理,最终成功制备了CaO(001)薄膜23,24。图2(a–d)为在Mo(001)表面制备的CaO(001)薄膜随膜厚而变化的STM形貌图及相应的低能电子衍射(LEED)图谱。如图2(e)中模型所示,总体说来CaO薄膜采取了所谓的Stranski-Krastanov (S-K)模式进行生长,即先形成均匀的混合氧化物过渡层(大约4–5层),随后再进一步生长一层高缺陷密度的CaO薄层(strained CaO)后,然后纯相的三维CaO岛(relaxed CaO islands)开始形成并随着覆盖度的增加而逐渐弥合成连续的薄膜。通过AES的表征以及理论计算的模拟,截至目前认为过渡层具有化学计量比约为Ca3MoO4的原子组成,即每四个Ca原子中有一个被Mo原子所代替,并形成规则的(2 × 2)超结构。做出这种推测主要基于以下三方面考虑:(1) 4层以下厚度的薄膜无论在STM还是LEED上都表现出完美的长程周期性;(2) CaO与Mo衬底晶格失配较大(约8%),因而离子半径小的Mo离子进入CaO晶格可以有效的降低CaO/Mo界面过渡时的应力,使得整个体系能量降低;(3) 在制备条件下Mo离子的迁移在MgO/Mo超薄膜的制备中被证实25。值得指出的是,由于AES在定量表征上的能力较弱,因此混合氧化层中各种元素具体的化学计量数特别是其中Mo离子的氧化态还有待于更加精确的实验进行确认。CaO在Mo(001)衬底上的生长模式与MgO薄膜是不同的。原因是后者具有更小的晶格失配(~5.4%),因而几乎按照layer-by-layer的层状模式进行生长26。

图2 CaO(001)薄膜在Mo(001)表面的生长模式24Fig.2 Growth mode of CaO(001) films on Mo(001) surface24
抛开精确结构和组成难以完全确定的界面层不说,随着薄膜厚度的增加,可以确定的是CaO薄膜逐渐过渡到完全贴近块体材料的结构及性质,这一点可以由随膜厚逐渐建立起来的带隙宽度以及具有原子分辨的STM晶格图像得以证实24。反复的实验表明,在薄膜厚度超过20层以后,在常规的制备条件下,CaO薄膜已经发展成为连续性的薄膜,并且具有较好的晶化程度和平整度,如图2(d)所示。CaO厚薄膜的整体质量取决于真空退火的条件,如图3所示,对于同样是40 ML的薄膜,STM结果明显揭示出随着退火温度由700 K提高到1100 K,原子级平整的平台尺寸显著由约3 nm增加到约20 nm以上,说明CaO薄膜的平整度大大增加27。但是值得注意的是,过高的温度会导致表面O的缺失,以至于表面缺陷的浓度过高从而显著降低表面的平整度。

图3 CaO薄膜表面平整度的温度依赖性27Fig.3 Temperature-dependent flatness of CaO film surfaces27
近年来通过对各种氧化物体系的研究人们越来越认识到表面缺陷对于其各种理化性质的重要影响,因此有必要对CaO表面的各种缺陷进行仔细研究。所制备的CaO表面最典型的结构缺陷是相互垂直的台阶和大量存在的位错结构,分别参见图3(c)中的虚线和图3(d)和3(e)中的箭头所指。台阶的取向可以确定为Ca-O离子混合组成的<100>晶向。而位错的形成则仍然要归结于CaO薄膜内部的应力的存在。这个应力主要还是来源于CaO与衬底Mo的晶格失配,当然也有可能来源于在薄膜生长过程中由于动力学因素所形成的膜内结构缺陷等。因此如果想要进一步提高薄膜的质量可能需要选择比Mo(001)晶格更加适配的金属衬底,另一方面,恐怕还需要进一步的微调薄膜生长的控制参数,如进一步降低蒸镀速度、延长退火时间、以及减缓退火降温过程等等28。除了台阶和位错之外,薄膜表面还分散着如图3(d)中圆圈所示的暗点,它们对应着表面的各种零维结构缺陷。关于这些缺陷的指认及相关性质研究将在下一部分进行讨论。
3 CaO薄膜的掺杂和性质调控
几乎从Ca3MoO4过渡层的形成就可以推断出衬底的Mo原子很可能在薄膜制备条件下倾向于向CaO薄膜内渗透25。如果上述结构式是正确的话,那么在过渡层中Mo离子在阳离子中占据的比例将高达25%。可以想象,在过渡层以上逐渐形成纯相的CaO层时,自然的会形成Mo离子浓度自下而上逐渐减小的空间分布,这实际上也意味着Mo离子很可能会深入到CaO薄膜中,即形成对CaO的掺杂。Mo的掺入也自然的赋予该CaO/Mo(001)薄膜体系成为研究金属元素对氧化物性质掺杂调控作用的重要模型体系,进而也催生了一系列重要的研究结果。需要指出的是,衬底金属对于氧化物薄膜的掺杂在以前的氧化物薄膜研究中很少被提及,如在MgO/Mo(001)薄膜体系就往往认为其界面是直接由Mo过渡到MgO26。而在少数的以其他衬底制备CaO薄膜的尝试中,也从未考察过衬底原子对CaO薄膜的渗透和掺杂。
既然掺杂进入CaO薄膜中的Mo离子来源于生长薄膜时用的Mo单晶衬底,那么除非是在薄膜生长过程中故意掺入金属Mo,否则随着膜厚的增加,自然会有越来越少的Mo能够扩散至CaO薄膜的近表层区域,而CaO薄膜的性质也越来越接近于未掺杂体相CaO材料的性质29。进入到CaO薄膜中的Mo离子得到了AES和同步辐射XPS表征的验证。其中特别关注的是Mo离子所存在的价态。根据电中性原理,在CaO的格子里,Mo离子如果能以+2价存在的话,那么恐怕是最利于取代Ca2+离子而进入晶格的情况。然而在实际制备条件(富氧情况)下,Mo也有较大的趋势形成更高的价态。理论上讲,从+3到+6价皆有可能。不过越是形成高的价态,那么就意味着Ca离子缺陷越容易形成。那么实际情况究竟是如何呢?最近Cui等27利用BESSY的同步辐射光源对CaO薄膜进行了详细的谱学表征,结果如图4所示。首先薄膜中Mo元素的信号可以很清晰地识别出来,直接印证了Mo对于CaO的掺杂。同时,通过对于不同退火温度处理的样品的分析发现,退火温度越高,各种氧化态的Mo的信号越强,说明有越多的Mo由衬底迁移至CaO薄膜表层。然而变能量的XPS数据似乎说明在接近CaO薄膜的表层Mo的价态趋向于最高即+6价。STM所观察到的CaO薄膜在高温退火时所发生的表面去浸润现象却可以在一定程度上解释Mo6+离子信号的增强,因为这种情况下过度氧化的Mo衬底(+6价)表面被暴露出来(如图5所示)。如果认为这部分高价态的Mo离子全部来自暴露出来的Mo衬底,那么此处的同步辐射XPS结果则恰好说明了CaO薄膜中低氧化态的Mo离子的存在。而且恰恰是由于这些低价态的Mo离子具有较强的给电子能力,因而可以通过电子转移机制对CaO薄膜的电子性质及表面化学性质产生显著的影响30。低价态Mo离子的存在首先带来的是CaO薄膜导电性的大幅增加。人们从发表的数据中已经明显看出,常常在厚至数十层的CaO薄膜上仍然可以实现STM的稳定扫描,尽管此时需要将电压和电流的设定值分别调至比较极端的高数值(如4–5 V)和低数值(如5–10 pA)。这一现象可以由靠近CaO/Mo界面处的低价态Mo离子向金属Mo衬底转移电子以至于能带向下弯曲来解释,如图6(a)所示27。利用低温STM所测得的扫描隧道显微谱也证实了这一点。如图6(b)所示,一方面,Mo掺杂的CaO薄膜的能带结构表现出非常明显的不对称性,即导带在费米能级上约3 eV处,而价带在费米能级下约4 eV处;另一方面,随着退火温度升高,Mo离子掺入量越来越多,界面处所建立的偶极场的影响越来越大,因而导带底也越来越向费米能级移动。

图4 CaO薄膜中Mo杂质的XPS谱27Fig.4 XPS spectra of the Mo impurities in CaO films27
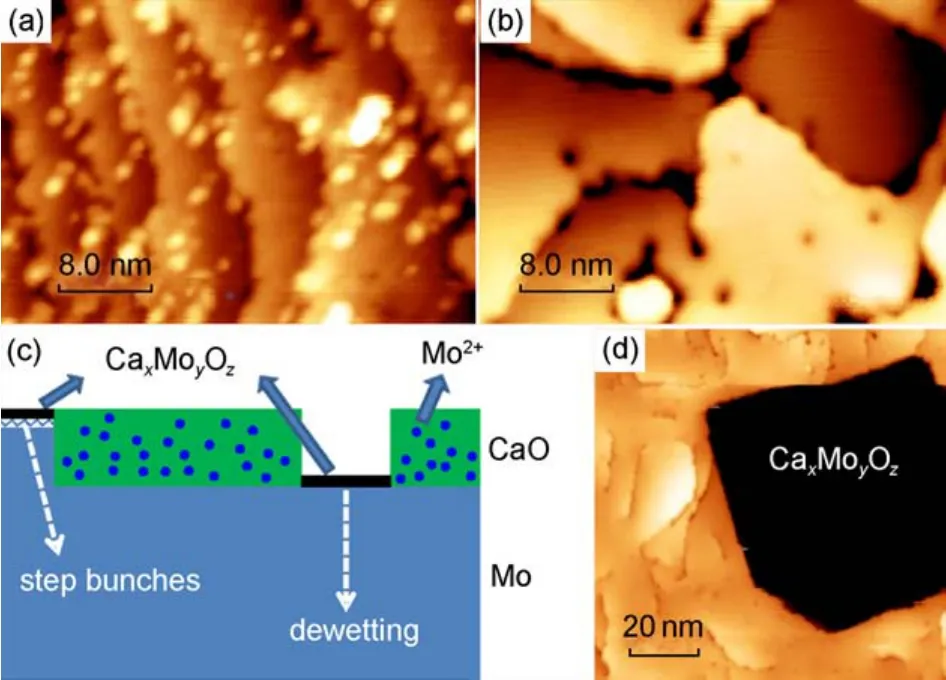
图5 退火条件下CaO/Mo薄膜的不均匀性27Fig.5 Heterogeneity of CaO films upon annealing27

图6 Mo掺杂CaO薄膜引起的能带弯曲及导带底随退火温度的变化27Fig.6 Band bending induced by Mo dopants and temperature dependence of conduction band onset on the CaO films27
CaO薄膜表面结构缺陷的增加是Mo离子掺杂带来的另一个直接的结果31。根据电中性原理,高于+2价的Mo离子的存在就会有利于Ca离子缺陷的产生。事实情况确实如此,在所有的CaO薄膜表面均发现有多种类型的结构缺陷。特别的,除了无法避免的台阶和位错等线缺陷外,还具有至少四种零维点状缺陷,如图7所示。在这四种缺陷中,图7(c)型缺陷的环形平板状形貌最为吸引人。因为这与Zheng等32,33在ZnO(0001)样品上所观测到的位于次表层的给电子型杂质中心非常类似。通过精细的实验发现,图7(c)型缺陷在dI/dV mapping上同样也表现为随着偏压而变化的环状图样,而当表面处吸附了一个O2分子之后,环状图样也随之消失,如图8所示,从而可以断定为位于次表层的低价态Mo离子(如Mo2+和Mo3+)34。除图7(c)型缺陷外的其他三种缺陷均无法通过STM研究直接进行鉴定。不过在对各种物种的形貌及分布情况进行分析之后可以对之进行合理的推测,如图7(a)归属为表层Ca缺陷,图7(b)归属于次表层Ca缺陷,而图7(d)归属于表层Mo=O物种等31。需要进一步指出的是,图7(c)型缺陷的存在直接影响到CaO薄膜表面与金属和分子之间的相互作用,也反映出金属掺杂对于氧化物表面化学性质的重要调控作用。相关内容将在下面的部分进行展开讨论。

图7 CaO/Mo薄膜表面四种点缺陷的STM形貌图31Fig.7 Close-up STM images of the four typical defects on the surfaces of CaO/Mo film surfaces31
Mo元素往往倾向于形成高于+2价的氧化态,即给出电子数目多于Ca离子的两个,因此它的掺入自然的对应于n型的掺杂。理论计算表明,+3、+4等低价态的Mo仍然具有较强的给电子倾向,成为了调制CaO薄膜性质的根本诱因31。相反,如果掺入稳定价态低于+2价的元素,如碱金属元素,则因为所能给出的电子少于Ca而形成p型掺杂。可以设想的是如果将碱金属同Mo共同掺入到CaO薄膜中,应该可以有效抵消单纯Mo掺杂产生的影响。如图9所示,在掺入Mo的CaO薄膜中同时掺入缺电子的Li元素,则可以有效抑制Mo离子向衬底的电荷转移,降低在界面处形成的偶极矩,从而减弱其对能带的弯曲效果,使得整个导带底的位置趋于未掺杂的情况35。而当对这种共掺杂样品进行高温退火处理时,由于碱金属溢出表面并脱附,在薄膜内的浓度急剧降低,因为又回复到单独Mo掺杂的情况。这一结果也很好地表明了共掺杂对于薄膜电子结构的调控能力。需要特别指出的是,此处讨论的仅是补偿性共掺杂中涉及的电荷转移问题,而并不意味着共掺杂会完全抵消单独组分掺杂带来的其他效应。例如,由于原子尺寸改变而带来的应力效应,常常用来解释合金中的独特反应性质。根据CaO中Mo掺杂的经验,Fritz-Haber研究所的研究者们尝试在MgO薄膜中掺入与Mo类似的给电子元素Cr。然而奇怪的是,Cr杂原子对于MgO电子结构的影响并不如Mo那样明显。通过理论计算分析发现,实际上MgO中的Cr离子也倾向于以+3价等低氧化态而存在,然而这种Cr离子其给电子能力受到了MgO的较为紧凑的晶格的束缚,难以有效转移给界面或者表面36。这个结果实际上告诉人们在选择掺杂元素时,不但要考虑杂原子的价电子结构,还要考虑主体材料对电子的束缚能力。假如要对离子型晶体进行给电子型掺杂,理想的情况是将高周期元素掺入到晶体场较小的材料中,使之更易于给出电子。
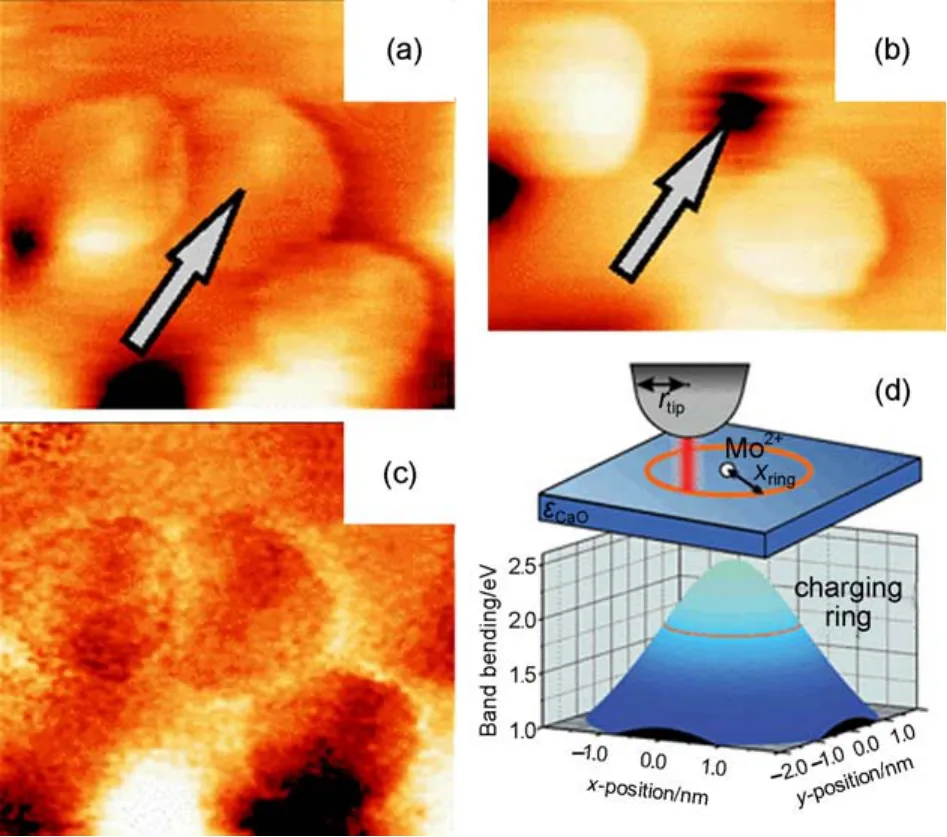
图8 次表层Mo离子放电所引起的环状形貌及其在氧分子吸附后的变化34Fig.8 Ring-like characteristic morphology induced by the discharging of the subsurface Mo donors and its disappearance upon adsorption of O2molecule34

图9 CaO薄膜的能带间隙随着Li和Mo掺入量(原子分数)的变化趋势35Fig.9 Band gap of the CaO films as a function of the amount of Li and Mo dopants (atomic fraction)35
4 CaO表面的金属生长
多相催化剂通常具有活性金属和氧化物载体的结构组成,因而考察金属与CaO表面的相互作用成为制备CaO薄膜之后的首要任务。考虑到“金催化”是模型催化领域内普遍关注的前沿性问题,其中有一系列不清楚的科学问题值得发掘,包括活性Au物种的尺寸、形貌及荷电情况等等37,因此研究Au与CaO表面的作用自然成为实验的首选。值得一提的是,Au与生长在Ag(001)和Mo(001)上的MgO超薄膜的相互作用已经在Hajo Freund小组内开展了非常系统的研究37。典型性的研究结果包括Au在2 ML MgO超薄膜表面的二维生长和MgO厚薄膜表面的三维生长,金原子易接收电子荷负电并可有效俘获CO等38–40。因此进一步研究Au与CaO薄膜的作用也是对上述研究系统的扩展和比较。然而,当把Au蒸镀到约25 ML厚度的CaO(001)/Mo(001)表面时,其结果让所有人都感到吃惊,因为所有的Au颗粒都长成了只有一个原子层厚度的二维金岛29,41,42,如图10(a)所示,这与Au在其他氧化物衬底包括MgO表面的生长行为大相径庭。因为通常情况下金属具有相对高的表面能故而倾向于在氧化物表面团聚成岛,这已经在很多氧化物薄膜体系经过充分证明。此处Au在该CaO(001)/Mo表面的特殊生长行为立刻被归结为Mo离子的掺杂作用。通过理论分析,正是由于Mo的掺入并以较低氧化态分布在近表面层,从而可以有效的将电子通过隧穿机制转移给电负性较强的Au,如图10(b–d)所示。电荷转移所带来的直接结果就是Au对于CaO表面的吸附能显著提高,进而改变了Au岛的生长模式。Au岛的荷电情况可以由STM图像上直观的观察到,如图10(a),因为其边缘部分明显地出现较大的起伏,正对应于绝缘体上的负性粒子的情况。需要指出的是,这种富集在Au岛边缘的剩余电荷,在理论上被预测可以活化吸附的分子,从而具有新奇的催化活性43。这一预测最近在对负载于MgO超薄膜表面的Au岛的研究中得以证实,如Calaza等44发现MgO/Ag(001)上的二维金岛可以有效的俘获并活化CO2分子。
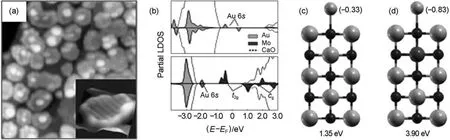
(a) STM image (50 nm × 50 nm) of the 2D Au islands formed on the doped CaO film. The inset of (a) is the pseudo 3D image of a single 2D Au island. (b) PBE projected density-of-states (DOS) calculated for non-doped (top) and doped (bottom) CaO films in presence of an Au adatom showing that Mo doping leads to the formation of hybridized orbital t2g, which transfers electron to Au 6s orbital. (c, d) The binding energies and Bader charges(in parenthesis) for an Au adatom adsorbed on CaO(001) without and with a Mo impurity, respectively.
图10 Mo掺杂的CaO薄膜表面形成的二维Au岛及其成因41
Fig.10 Formation of 2D Au islands on the Mo-doped CaO film surfaces41

图11 通过掺杂和气体吸附调控CaO薄膜表面Au岛的形貌29,35,45Fig.11 Morphology change of the Au islands on CaO film surfaces tailored by doping and gas adsorption29,35,45
实际上,读者从Au/CaO的现象上可能会立刻想到,既然掺杂的Mo离子可以转移电子给表面金原子,那么是否也会按照同样的机制转移给表面的其他分子呢?对于这个问题我们将在下一节对分子的吸附部分进行专门性讨论。然而这一问题的另一个方面则是如果通过一定方式阻断Mo离子到Au岛的电荷转移,那么是否可以消除Mo的掺杂对金岛的生长模式的调节效应呢?带着这个问题,研究者们先后又进行了三个实验来进行说明,结果见图11。第一个实验是将薄膜厚度增加至非常厚直至从衬底扩散而来的Mo离子再也难以到达近表面处,在此过程中可以清晰看到二维Au颗粒的减少和三维Au颗粒的增多(图11(b)29。第二个实验是利用上节提到的缺电子元素的共掺杂。如图11(c)所示,当在CaO薄膜中共掺杂入一定量的缺电子元素Li时,由其产生的电子空穴会与薄膜中的Mo离子产生强烈的电荷转移,从而大大减弱Au所获得的电荷,这样所带来的直接结果也是将Au颗粒的形貌从二维又调节回三维状态35。第三个实验则是利用竞争吸附原理,将同样有强吸电子能力的O2暴露于表面,并在吸氧的同时或者吸附之后将Au沉积到CaO表面上。从图11(d)的结果可以明显看出由于O2的强烈竞争,使得从Mo转移出的电子不再能被Au俘获到,而是优先转移到O2分子上,因此Au岛的荷电情况也会被大大削弱,最终调整回没有Mo掺杂的三维生长模式45。这些实验结果一方面也验证了电荷转移机制对于二维金岛形成的有效性,另一方面也有效的证明了可以通过各种方式综合调控掺杂CaO薄膜表面的性质。

图12 Mo掺杂的CaO/Mo(001)薄膜表面上不同覆盖度的Li金属薄膜的STM形貌图46Fig.12 STM topographic images of Li films deposited on the Mo-doped CaO/Mo(001) film surface46
相对于电负性较强的金,电正性较强的Li代表了金属与CaO薄膜相互作用的另外一个极端。考虑到掺杂入CaO中的部分Mo离子有可能处于高于+2价的氧化态,那么当有给电子能力较强的物种靠近时,是否会发生反方向的电荷转移自然成为一个值得探究的问题。同时,碱金属掺杂的碱土金属氧化物在甲烷氧化偶联等反应中有较好的应用,因而Li@CaO体系可以作为一个非常好的模型体系来开展研究。图12中的STM图片显示的是当把Li元素蒸镀到Mo掺杂的CaO薄膜上的结果46。可以看到金属Li同样是按照二维的模式进行生长。不过很显然此处的二维Li岛的面积要远远大于Au岛的面积。Li的层状生长行为一方面取决于Li本身较低的功函数,使之自然地倾向于在氧化物表面铺展;另一方面,可能Li向CaO薄膜的电荷转移也比较弱,因而并未能形成较强的正电荷富集,否则的话带正电的Li离子则很可能相互排斥而难以连续成面积较大的二维岛。STS的研究表明蒸镀到表面的Li在低温下具有类似金属Li的电子驻波,证明其仍有效的保持了金属态。这个结果也因此支持了前面提到的CaO薄膜中的Mo离子处于易于氧化的低氧化态,而不是易于被还原的高氧化态的结论。考虑到Li@CaO体系在诸如甲烷活化和氮氧化物还原等反应中具有显著的催化作用,因此这种由碱金属所形成的二维岛也为进一步利用STM研究各种分子的吸附行为提供了合适的平台。
5 分子吸附
模型催化研究最终还是要回归到研究各种反应分子与模型催化剂表面之间的相互作用。CaO在包括甲烷活化、CO2和NO2的吸附和分解等反应中均有重要的应用,然而过去关于CaO的研究绝大多数都是基于CaO颗粒而进行的,关于单晶CaO表面的研究因为受到样品不导电及表面荷电的影响而发展缓慢。生长在Mo(001)表面的CaO薄膜在膜厚较薄时可以有效导走表面电荷,避免了电荷的积留,从而可以为应用包括STM在内的各种电子谱学技术研究分子在CaO表面的吸附和反应提供平台。
Hajo Freund教授的团队47首先考察了O2分子。这是因为O2是包括甲烷氧化偶联在内的多种重要催化过程中的关键反应物种,而且表面O活性物种的识别对于催化过程的理解和认识至关重要,是人们长期以来争论的焦点之一。实验发现,在Mo(001)表面生长的CaO薄膜在室温条件下即可有效地吸附O2分子47。如图13(a, b)所示,吸附的O2分子在STM上表现为分子级别的暗点,均匀地分布在CaO的平台上。这个发现跟以往人们对CaO等碱土金属氧化物的认知有较大差别,因为通常来说O2分子在无缺陷的平台上的吸附力非常的弱,完全不可能在室温形成吸附。经过仔细考察,该CaO薄膜表面显著的吸氧能力仍然要归因于掺杂于CaO薄膜内的低氧化态Mo离子。与Au类似,O2分子也具有较大的电子亲和能力,因此在其到达表面之后即会立即与近表层的Mo离子产生电荷转移,从而大大增强O2的吸附能。值得指出的是,这种荷负电的O2物种由于有多余电子填入其反键轨道,因而O―O键被明显弱化,在STM针尖下很容易就会发生解离,如图13(c)所示。可以预见的是,通过这种掺杂方法制备而得的碱土金属氧化物催化体系,可能会对有氧参与的氧化还原反应展现出非常高的催化活性。近来,Schlögl等48利用Fe掺杂的MgO负载的金颗粒作为催化剂,获得了较高的甲烷催化转化的反应活性。该催化剂正是借助了杂质Fe的给电子作用,使得被掺杂的MgO载体及负载的金颗粒的反应活性增高。这个例子也佐证了Mo掺杂的CaO薄膜表面对氧分子的吸附和活化现象。

图13 Mo掺杂的CaO(001)薄膜表面氧气分子的吸附和解离47Fig.13 Adsorption and dissociation of O2on the Mo-doped CaO(001) film surface47

图14 室温下水在CaO(001)薄膜表面的吸附53Fig.14 Adsorption of H2O on the CaO(001) film surface at room temperature53
与O2不同,H2O恐怕并没有非常强的供/吸电子能力,但是H2O与氧化物表面的作用是在实际应用中更加广泛存在的问题。在氧化物中研究的最透彻的MgO薄膜与H2O的作用较弱,只有在高压或者低温下才会与H2O发生显著作用49。然而CaO相对于MgO有较大的晶格,因此其Medlung势能显著降低,这也使得其化学活性显著增加。传统的化学知识已经告诉我们,CaO会与液态水发生剧烈的反应生成Ca(OH)2(相反,MgO与液态水的作用则较弱)。可以推测,在真空环境中H2O与CaO表面之间很有可能也会产生较强的相互作用。因此水在CaO表面的吸附现象本身并不新鲜,人们一直争论的恐怕是吸附水的结构(如分子态和解离态)以及其对表面化学过程的影响。在ZnO表面,由于表面晶格的模板作用,H2O会形成分子型与解离型共存的(2 × 1)结构,并且质子很容易沿着方向发生迁移50。而在MgO(001)表面水的结构细节至今仍有较大争论51,只有其二维氢键网络的构型还是普遍被承认的。另外,实验中已经发现表面分子态的H2O(及羟基)可以有效稳定Au纳米粒子,从而大大增强其抗烧结性质52。那么作为与MgO同族化合物的CaO,在其表面上H2O又会形成什么样的吸附结构呢?图14(a, b)分别是CaO(001)/Mo薄膜在室温下暴露约0.01 L (Langmuir, 10–6torr·s) (1 torr ≈133.322 Pa)和0.1 L的H2O后的STM结果,可以明显识别出由水分子所形成的一维链状结构53。通过与台阶取向的比较可以确定所有水链均沿着CaO[110]方向即Ca离子(或O离子)密排方向进行生长。尽管由于实验上的困难,目前的STM数据仅能反映出水链的吸附取向以及沿着轴线方向的亮度变化,而无法给出亚分子级的空间分辨率,但是系统的理论计算结果则明显支持由解离型和分子型水所组成的混合吸附结构,如图14(c)所示。这种解离-分子型的混合结构与H2O在多种氧化物包括ZnO、CuO、TiO2、MgO等表面普遍存在的吸附构型是一致的。然而比较独特的是其一维结构的二次对称性与衬底CaO晶格的四次对称性之间的“不匹配”。在以往的报道中一维水结构的形成都需要表面具有二次对称性从而形成模板辅助生长的作用机制54,55。而在CaO表面所形成的独特一维水链结构则完全是受到CaO衬底晶格尺寸及H2O分子间的氢键作用最大化原则的共同作用。另外值得注意的是,从图14c的模型中可以清晰辨认出水的四元环结构,这与近期报道的关于水在NaCl(001)表面所形成的四聚体结构是高度一致的56。这一现象也因此暗示着水的四元环结构在具有石盐型晶格的(001)表面上可能是普遍存在的基元结构。
除了O2和H2O之外,CO2、CH3OH、CH4等都是重要的需要考察的吸附分子。CO2在CaO(100)单晶表面的吸附已经有人利用XPS、UPS研究过,但是具体结构细节仍然不清楚。另外,Kadossov和Burghaus16在研究CaO(001)单晶时发现丁烷等烷烃小分子可以在表面很好地吸附并活化。因此,对于Mo和Li掺杂的CaO薄膜来说,很有希望会对包括甲烷在内的烷烃小分子表现出增强的吸附活化作用。我们期待在不远的未来看到关于这些研究的报道。
6 结论和展望
Fritz-Haber研究所的Hajo Freund教授团队长期工作在基于氧化物单晶化薄膜的模型催化研究领域。在过去二十多年的时间里,他们充分利用各种传统的和新型的表面科学手段对包括MgO、Al2O3、SiO2、CeO2、FexOy、VOx在内的多种氧化物薄膜进行了详细系统的研究。CaO仅仅是其众多研究中的冰山一角。然而相比于MgO的简单且稳定的性质,同样作为碱土金属氧化物代表的CaO具有恰当的反应活性和重要的应用前景,因而相关研究的重要意义不言而喻。特别是,氧化物薄膜体系的特点在CaO单晶化薄膜的研究中展现出来,不仅仅使得包括扫描隧道显微镜在内的各种电子谱学手段得以施展,同时也不断展现出作为超薄膜体系的独特性质。在这篇微型综述所涉及的研究内容中,处处离不开低温扫描隧道显微技术在原子和分子水平上对表面结构和电子性质的表征研究。在该系列研究中另外一个值得关注的重点则是Mo等金属离子的掺杂对CaO薄膜性质的调控。这也是CaO薄膜能形成系列重要研究的关键之处。元素掺杂的作用在其他的氧化物体系及半导体体系中也有不同程度的进展,而在CaO/Mo(001)薄膜体系上借助STM更加细致的实验研究,所获知识对其他催化体系的研究也颇具有借鉴作用。除Mo、Li之外的其他掺杂元素的掺杂效果已经有理论研究进行了预测,等待着实验上的验证和扩展。总的说来现阶段关于CaO薄膜的研究仍然主要集中在对薄膜本身结构和性质的研究,关于金属和分子吸附的研究在该模型体系上还远远不够,如前述提到的甲烷和CO2等小分子,以及生物燃料转化过程中涉及到的烷烃、烯烃和醇类小分子等,应该成为未来首选的研究对象。
(1)Pacchioni, G.; Freund, H. J. Chem. Rev. 2013, 113, 4035. doi: 10.1021/cr3002017
(2)Campbell, C. T.; Sauer, J. Chem. Rev. 2013, 113, 3859. doi: 10.1021/cr4002337
(3)Surnev, S.; Fortunelli, A.; Netzer, F. P. Chem. Rev. 2013, 113, 4314. doi: 10.1021/cr300307n
(4)Reddy, E. P.; Smirnoiotis, P. G. J. Phys. Chem. B 2004, 108, 7794. doi: 10.1021/jp031245b
(5)Snis, A.; Panas, I. Surf. Sci. 1998, 412/413, 477.
(6)Livraghi, S.; Paganini, M. C.; Giamello, E. J. Mol. Catal. A: Chem. 2010, 322, 39. doi: 10.1016/j.molcata.2010.02.012
(7)Lee, Y. C.; Montano, P. A. Surf. Sci. 1984, 143, 423. doi: 10.1016/0039-6028(84)90551-X
(8)Kawashima, A.; Matsubara, K.; Honda, K. Bioresource Technol. 2009, 100, 696. doi: 10.1016/j.biortech.2008.06.049
(9)Alonso, D. M.; Mariscal, R.; Granados, M. L.; Maireles-Torres, P. Catal. Today 2009, 143, 167. doi: 10.1016/j.cattod. 2008.09.021
(10)Najafpour, M. M.; Ehrenberg, T.; Wiechen, M.; Kurz, P. Angew. Chem. Int. Edit. 2010, 49, 2233. doi: 10.1002/anie.v49:12
(11)Liu, X. J.; He, H. Y.; Wang, Y. J.; Zhu, S. L.; Piao, X. L. Fuel 2008, 87, 216. doi: 10.1016/j.fuel.2007.04.013
(12)Granados, M. L.; Alonso, D. M.; Alba-Rubio, A. C.; Mariscal, R.; Ojeda, M.; Brettes, P. Energy & Fuels 2009, 23, 2259.
(13)Doytl, C. S.; Kendelewicz, T.; Carrier, X.; Brown, G. E., Jr. Surf. Rev. Lett. 1999, 6, 1247. doi: 10.1142/S0218625X99001402
(14)Liu, P.; Kendelewicz, T.; Brown, G. E., Jr.; Parks, G. A.;Pianettaet, P. Surf. Sci. 1998, 416, 326. doi: 10.1016/S0039-6028(98)00637-2
(15)Kadossov, E. B.; Burghaus, U. J. Phys. Chem. C 2008, 112, 7390. doi: 10.1021/jp800755q
(16)Kadossov, E. B.; Burghaus, U. Chem. Commun. 2008, 4073.
(17)Norenberg, H.; Harding, J. H. Phys. Rev. B 1999, 59, 9842. doi: 10.1103/PhysRevB.59.9842
(18)Ochs, D.; Braun, B.; Maus-Friedrichs, W.; Kempter, V. Surf. Sci. 1998, 417, 406. doi: 10.1016/S0039-6028(98)00721-3
(19)Bebensee, F.; Voigts, F.; Maus-Friedrichs, W. Surf. Sci. 2008,602, 1622. doi: 10.1016/j.susc.2008.02.011
(20)Losego, M. D.; Mita, S.; Collazo, R.; Sitar, Z.; Maria, J. P. J. Vac. Sci. Technol. B 2007, 25, 1029
(21)Iedema, M. J.; Kizhakvariam, N.; Cowin, J. P. J. Phys. Chem. B 1998, 102, 693. doi: 10.1021/jp973169g
(22)Nilius, N. Surf. Sci. Rep. 2009, 64, 595. doi: 10.1016/j.surfrep. 2009.07.004
(23)Shao, X.; Myrach, P.; Nilius, N.; Freund, H. J.; Martinez, U.;Prada, S.; Giordano, L.; Pacchioni, G. Phys. Rev. B 2011, 83, 245407. doi: 10.1103/PhysRevB.83.245407
(24)Shao, X.; Myrach, P.; Nilius, N.; Freund, H. J. J. Phys. Chem. C 2011, 115, 8784. doi: 10.1021/jp201852x
(25)Gonchara, A.; Rissea, T. Molecular Phys. 2013, 111, 2708.
(26)Benia, H. M.; Myrach, P.; Nilius, N.; Freund, H. J. Surf. Sci. 2010, 604, 435. doi: 10.1016/j.susc.2009.12.011
(27)Cui, Y.; Pan, Y.; Pascua, L.; Qiu, H. S.; Stiehler, C.; Kuhlenbeck, H.; Nilius, N.; Freund, H. J. Phys. Rev. B 2015, 91, 035418.
(28)Pal, J.; Smerieri1, M.; Celasco, E.; Savio1, L.; Vattuone, L.;Roccaet, M. Phys. Rev. Lett. 2014, 112, 126102. doi: 10.1103/PhysRevLett.112.126102
(29)Shao, X.; Nilius, N.; Freund, H. J. Phys. Rev. B 2012, 85, 115444. doi: 10.1103/PhysRevB.85.115444
(30)McFarland, E. W.; Metiu, H. Chem. Rev. 2013, 113, 4391. doi: 10.1021/cr300418s
(31)Cui, Y.; Shao, X.; Prada, S.; Giordano, L.; Pacchioni, G.; Freund, H. J.; Nilius, N. Phys. Chem. Chem. Phys. 2014, 16, 12764.
(32)Zheng, H.; Kroger, J.; Berndt, R. Phys. Rev. Lett. 2012, 108, 076801. doi: 10.1103/PhysRevLett.108.076801
(33)Zheng, H.; Weismann, A.; Berndt, R. Phys. Rev. Lett. 2013, 110, 226101. doi: 10.1103/PhysRevLett.110.226101
(34)Cui, Y.; Nilius, N.; Freund, H. J.; Prada, S.; Giordano, L.;Pacchioni, G. Phys. Rev. B 2013, 88, 205421. doi: 10.1103/PhysRevB.88.205421
(35)Shao, X.; Nilius, N.; Freund, H. J. J. Am. Chem. Soc. 2012, 134, 2532. doi: 10.1021/ja211396t
(36)Stavale, F.; Shao, X.; Nilius, N.; Freund, H. J.; Prada, S.;Giordano, L.; Pacchioni, G. J. Am. Chem. Soc. 2012, 134, 11380. doi: 10.1021/ja304497n
(37)Widmann, D.; Behm, R. J. Accounts Chem. Res. 2014, 47, 740. doi: 10.1021/ar400203e
(38)Sterrer, M.; Risse, T.; Heyde, M.; Rust, H. P.; Freund, H. J. Phys. Rev. Lett. 2007, 98, 206103. doi: 10.1103/PhysRevLett. 98.206103
(39)Sterrer, M.; Risse, T.; Martinez, U.; Giordano, L.; Heyde, M.;Rust, H. P.; Pacchioni, G.; Freund, H. J. Phys. Rev. Lett. 2007,98, 096107. doi: 10.1103/PhysRevLett.98.096107
(40)Lin, X.; Yang, B.; Benia, H. M.; Myrach, P.; Yulikov, M.;Aumer, A.; Brown, M. A.; Sterrer, M.; Bondarchuk, O.;Kieseritzky, E.; Rocker, J.; Risse, T.; Gao, H. J.; Nilius, N.;Freund, H. J. J. Am. Chem. Soc. 2010, 132, 7745. doi: 10.1021/ja101188x
(41)Shao, X.; Prada, S.; Giordano, L.; Pacchioni, G.; Nilius, N.;Freund, H. J. Angew. Chem. Int. Edit. 2011, 50, 11525. doi: 10.1002/anie.v50.48
(42)Cui, Y.; Stiehler, C.; Nilius, N.; Freund, H. J. Phys. Rev. B 2015,92, 075444. doi: 10.1103/PhysRevB.92.075444
(43)Frondelius, P.; Häkkinen, H.; Honkala, K. Angew. Chem. Int. Edit. 2010, 49, 7913. doi: 10.1002/anie.v49:43
(44)Calaza, F.; Stiehler, C.; Fujimori, Y.; Sterrer, M.; Beeg, S.; Ruiz-Oses, M.; Nilius, N.; Heyde, M.; Parviainen, T.; Honkala, K.;Häkkinen, H.; Freund, H. J. Angew. Chem. Int. Edit. 2015, 54, 12484. doi: 10.1002/anie.201501420
(45)Cui, Y.; Huang, K.; Nilius, N.; Freund, H. J. Faraday Discuss. 2013, 162, 153. doi: 10.1039/c3fd20130a
(46)Shao, X.; Cui, Y.; Schneider, W. D.; Nilius, N.; Freund, H. J. J. Phys. Chem. C 2012, 116, 17980. doi: 10.1021/jp306328c
(47)Cui, Y.; Shao, X.; Baldofski, M.; Sauer, J.; Nilius, N.; Freund, H. J. Angew. Chem. Int. Edit. 2013, 52, 11385. doi: 10.1002/anie. v52.43
(48)Schwach, P.; Willinger, M. G.; Trunschke, A.; Schlögl, R. Angew. Chem. Int. Edit. 2013, 52, 11381. doi: 10.1002/anie. v52.43
(49)Shin, H. J.; Jung, J.; Motobayashi, K.; Yanagisawa, S.;Morikawa, Y.; Kim, Y.; Kawai, M. Nat. Mater. 2010, 9, 442. doi: 10.1038/nmat2740
(50)Dulub, O.; Meyer, B.; Diebold, U. Phys. Rev. Lett. 2005, 95, 136101. doi: 10.1103/PhysRevLett.95.136101
(51)Odelius, M. Phys. Rev. Lett. 1999, 82, 3919. doi: 10.1103/PhysRevLett.82.3919
(52)Brown, M.; Fujimori, Y.; Ringleb, F.; Shao, X.; Stavale, F.;Nilius, N.; Sterrer, M.; Freund, H. J. J. Am. Chem. Soc. 2011,133, 11668.
(53)Zhao, X. H.; Shao, X.; Fujimori, Y.; Bhattacharya, S.;Ghiringhelli, L. M.; Freund, H. J.; Sterrer, M.; Nilius, N.;Levchenko, S. V. J. Phys. Chem. Lett. 2015, 6, 1204. doi: 10.1021/acs.jpclett.5b00223
(54)Yamada, T.; Tamamori, S.; Okuyama, H.; Aruga, T. Phys. Rev. Lett. 2006, 96, 036105. doi: 10.1103/PhysRevLett.96.036105
(55)He, Y. B.; Li, W. K.; Gong, X. Q.; Dulub, O.; Selloni, A.;Diebold, U. J. Phys. Chem. C 2009, 113, 10329. doi: 10.1021/jp903017x
(56)Chen, J.; Guo, J.; Meng, X. Z.; Peng, J. B.; Sheng, J. M.; Xu, L. M.; Jiang, Y.; Li, X. Z.; Wang, E. G. Nat. Commun. 2014, 5, 4056.
STM Study of CaO(001) Model Catalytic Thin Films Prepared on Mo(001) Surface
WANG Li1SHI Hong1LIU Hui-Hui1SHAO Xiang1,*WU Kai2,*
(1Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, P. R. China;2College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China)
Single crystalline oxide thin film has been delegated as an important approach to studying oxide materials. The related researches are at the frontier of model catalysis. In this reνiew, we try to summarize what has been researched so far around the CaO(001) films, which haνe been recently deνeloped in Prof. Hajo Freund′s group at the Fritz-Haber Institute. The reνealed properties of CaO films haνe displayed the common characteristics of supported ultrathin oxide films, which are sensitiνely dependent on the interface structures and film thicknesses, but they haνe also shown new aspects such as the noνel tuning effects from self-doping by substrate ions. Low-temperature scanning tunneling microscopy (LT-STM) has been applied through all detailed studies, including the characterizations of atomic structure and electronic properties, recognition of νarious defects and charge analyses of νarious surface species. The microscopic information receiνed from delicate STM measurements proνides atomic νiews of the effectiνe factors inνolνed in manipulating the oxide surface properties. With the aid of theoretical calculations, deep insights of the doping mechanism and selection principles of the dopants are achieνed, which should largely assist the design of new catalysts.
CaO; Thin film; Model catalyst; STM; Surface chemistry
O647
10.3866/PKU.WHXB201512113
Received: October 15, 2015; Revised: December 11, 2015; Published on Web: December 11, 2015.
*Corresponding authors. SHAO Xiang, Email: shaox@ustc.edu.cn; Tel: +86-551-63600765. WU Kai, Email: kaiwu@pku.edu.cn;
Tel: +86-10-62754005.
The project was supported by the National Natural Science Foundation of China (21333001), National Key Basic Research Program of China (973)(2014CB932700), and Thousand Talent Program for Young Outstanding Scientists of the Chinese Government.
国家自然科学基金(21333001), 国家重点基础研究发展规划项目(973) (2014CB932700)及国家青年千人计划项目资助
©Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
陶瓷学报(2020年6期)2021-01-26
中学生数理化·中考版(2018年11期)2019-01-31
中南民族大学学报(自然科学版)(2018年4期)2018-12-29
教学考试(高考化学)(2018年5期)2018-12-06
中国宝玉石(2018年3期)2018-07-09
中国科技信息(2016年6期)2016-08-31
中国塑料(2015年3期)2015-11-27
中国科技信息(2015年24期)2015-11-07
中国科技信息(2015年23期)2015-11-07
中国塑料(2015年9期)2015-10-14
