
新型吡啶氮芥类化合物的合成及其抗微生物活性研究
2016-11-11 02:08曹守莹白长存陶兆林梁丽丽
安徽医科大学学报 2016年8期
曹守莹,白长存,陶兆林,梁丽丽
新型吡啶氮芥类化合物的合成及其抗微生物活性研究
曹守莹1,白长存2,陶兆林1,梁丽丽
目的 以2,6-二甲基吡啶为起始原料,经氧化、酯化、还原、氯化、N-烷基化等多步反应合成了吡啶双氮芥类化合物及其盐酸盐、硝酸盐。方法 目标化合物的结构经1H NMR、IR、MS和元素分析证实。结果 体外抗菌、抗真菌活性研究表明:所测试的大多数化合物均可有效抑制被测微生物的生长,且目标化合物对金黄色葡萄球菌表现出很强的专一选择性抑制作用,活性与临床药物相当或优于临床药物。其中吡啶双氮芥化合物7、8a和8b对金黄色葡萄球菌的选择性抑制作用显著,与参照药物氯霉素相当,吡啶双酰胺氮芥化合物11对金黄色葡萄球菌的抑制能力是氯霉素的两倍。结论 此类化合物有望作为潜在的抗金黄色葡萄球菌先导化合物,值得进一步研究其构效关系。实验培养得到了中间体产物6的晶体结构,单晶X射线衍射测试表明6的晶体结构属正交晶系P21212空间群。
氮芥;吡啶;抗细菌;抗真菌
氮芥类药物是一类有β-氯乙胺结构的化合物,因有广泛的生物活性和重要的药理活性如抗细菌[1]、抗真菌[2]、抗肿瘤[3-5]、抗病毒[6]等而备受关注。氮芥类药物也是临床上应用较早的一类重要的化学合成抗癌药,迄今为止仍被用于治疗各类恶性肿瘤,在抗癌药物中占有重要的地位[7]。近年来随着研究的深入,结构修饰后的氮芥类化合物也呈现出优异的抗微生物活性[2],在医药领域显示出宽广的应用前景,其相关研究与开发也渐趋活跃、发展迅速。吡啶及其衍生物作为一种重要的芳氮杂环类化合物,在多种天然产物的母体结构中广泛存在。吡啶类化合物还是化学工业,特别是精细化工行业的重要原料[8],应用范围涉及医药中间体、农药等多个领域,而在医药领域,如抗菌、抗癌、抗病毒等方面也发挥着重要作用[9-11]。研究[12-13]表明,在药物结构中引入吡啶环可以有效地调节药物的脂水分配系数和酸碱平衡常数。近年来,将氮芥类化合物的烷基化部分和吡啶环构筑于同一分子结构中的化合物鲜见文献报道。因此基于国际、国内抗微生物药物研发的新趋势,该研究采用药物设计活性基团拼合原理,将氮芥类药物的药效团β-氯乙胺和吡啶环构筑于同一分子结构中,首次设计合成了几个结构新颖的吡啶氮芥类化合物,并进行了抗微生物活性测试,对其构效关系作了初步研究。合成路线如图1所示,所合成的目标化合物均未被文献报道。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器 熔点仪:X-6型精密显微熔点测定仪(温度未校正);Bluker SMART APEXⅡ-CCD X射线单晶衍射仪;红外光谱仪:Bio-Rad FTS-185,Bruker RFS100/S(KBr压片);核磁共振仪:Bruker AV 300核磁共振仪(TMS内标);质谱仪:LCMS-2010A(ESI电离源);元素分析仪:Carlo Erba1106型元素分析仪。
1.1.2 试剂 甲醇、乙腈、THF等试剂均为市售分析纯;硅胶:青岛海洋化工厂200-300目、300-400目;TLC薄层层析硅胶板HSGF 254型;2、6-二甲基吡啶、二乙醇胺(成都科龙化工有限公司);氟康唑、氯霉素(珠海嘉成生物科技有限公司);牛肉膏、蛋白胨、葡萄糖、吗啡啉丙磺酸、琼脂、酵母浸膏等(北京奥博星生物技术有限公司);RMPI 1640(美国Gibco BRL公司)。
1.1.3 体外抗微生物活性测试用细菌和真菌 体外抗微生物活性测试的细菌包括3株革兰阳性菌:金黄色葡萄球菌(Staphylococcus aureus ATCC 6538)、耐甲氧西林金葡菌(MRSA N 315)和枯草芽孢杆菌(Bacillus subtilis ATCC 21216);3株革兰阴性菌:大肠杆菌(Escherichia coli ATCC 8099)、铜绿假单胞菌(Pseudomonas aeruginosa ATCC 27853)和变形杆菌(Bacillus proteus ATCC 13315);2株真菌:白色念珠菌(Candida albicans ATCC 76615)和假丝酵母菌(Candida mycoderma ATCC 96918)。
1.2 合成及结构表征
1.2.1 中间体2,6-吡啶二甲酸(2)的合成 把2,6-二甲基吡啶1(10.7 g,0.1 mol)溶于500 ml水中,17 h内分8次加入KMnO4(41.6 g,5.3 mol)。待反应原料2,6-二甲基吡啶反应完毕后,过滤,滤液浓缩至150 ml,加入30 ml浓盐酸。将混合液加热至沸腾使浓缩出来的固体充分溶解,然后慢慢使其自然冷却到室温,在5℃条件下低温保存过夜,2,6-吡啶二甲酸慢慢析出。过滤混合液,得到2,6-吡啶二甲酸的白色固体,用30 ml冷水洗涤固体,空气干燥,收率62%,m.p.255~257℃。
1.2.2 中间体2,6-吡啶二甲酸甲酯(3)的合成
2,6-吡啶二甲酸2(10.1 g,0.06 mol)和50 ml SOCl2置于150 ml烧瓶中,加热回流10 h,剩余SOCl2蒸馏除去。冰浴下冷却残留物10 min,撤去干燥管,滴液漏斗缓慢逐滴滴入250 ml无水甲醇,撤去冰浴,加热使反应体系回流反应30 min,停止反应,蒸馏去除150 ml甲醇,冰浴冷却反应体系,化合物2,6-吡啶二甲酸甲酯3析出,继续用甲醇洗去其上杂质,收率93.7%,m.p.119~122℃。
1.2.3 中间体2,6-二羟甲基吡啶(4)的合成 2,6-
吡啶二甲酸甲酯3(11.3 g,0.06 mol)和250 ml无水乙醇置于50 ml烧瓶中,冰浴下搅拌冷却15 min,撤去干燥管,分数次缓慢加入硼氢化钠(13 g,0.35 mol),加上干燥管,0℃继续反应1 h。撤去冰浴,加热使反应体系升温至251℃,反应3 h,继续加热至回流反应10 h,停止反应。蒸馏去除溶剂后加入100 ml丙酮,继续加热回流1 h,停止反应,减压蒸馏去除溶剂,将残留物溶解于400 ml水中,用氯仿萃取水溶液12 h,得到化合物2,6-羟甲基吡啶4的白色固体,收率66%,m.p.117~119℃。
1.2.4 中间体2,6-二氯甲基吡啶(5)的合成 氮气保护下,在250 ml圆底烧瓶中加入新蒸的干燥四氢呋喃(100 ml)和化合物4(8.79 g,0.06 mol),冰浴搅拌下冷却10 min,用滴液漏斗缓慢逐滴滴入15 ml SOCl2,0℃继续反应30 min。撤掉冰浴,反应体系缓慢升温至室温后加热回流8 h,停止反应。加入甲苯共沸除去过量SOCl2,得到产物2,6-二氯甲基吡啶的白色盐酸盐固体。把5的盐酸盐固体溶解于200 ml水中,加入饱和NaHCO3溶液调pH至8,化合物5析出,过滤水洗,用石油醚重结晶得到无色针状纯净的2,6-二氯甲基吡啶5,收率53%,m.p.74~75℃。
1.2.5 中间体2,2′,2″,2‴{吡啶-2,6-二取代[双(亚甲基)双(脲三基)]}四醇(6)的合成 在100 ml圆底烧瓶中加入乙腈(15 ml)、化合物5(1.76 g,0.01 mol)和二乙醇胺(3.16 g,0.03 mol),混合均匀后50℃搅拌过夜反应,TLC跟踪监测至反应完毕,停止反应。冷却反应体系,反应混合液旋蒸去溶剂,残留物加入饱和NaHCO3溶液洗涤,氯仿萃取,有机层用MgSO4干燥,旋蒸去溶剂乙腈,残余物经硅胶柱色谱,得目标产物白色固体,收率95%,m.p. 45~47℃。IR(KBr):ν 3389(OH),3009(Ar-H),2933,2887,2831,1592,1577,1446,1385,1338,1156,824,464 cm-1;1H NMR(CDCl3,300 MHz):δ 7.59~7.64(t,1H,J=15 Hz,Py 4-H),7.06~7.09(d,2H,J=9 Hz,Py 3-H,5-H),3.76(s,4H,PyCH2N),3.60~3.63(t,8H,J=9 Hz,CH2OH),2.68~2.72(t,8H,J=12 Hz,NCH2)ppm;13C NMR(CDCl3,75 MHz)δ:161(pyridine 2-C),137(thienyl 4-C),122(thienyl 3-C),63(pyridine-CH2),59(NCH2),41.5(CH2Cl)ppm。
1.2.6 目标化合物N,N′-[吡啶-2,6-二取代双(亚甲基)]双[2-氯-N-(2-氯乙基)乙胺](7)的合成[14]
氮气保护下,在150 ml圆底烧瓶中加入新蒸的干燥四氢呋喃(50 ml)和化合物6(1.3 g,0.04 mol),冰浴搅拌下冷却10 min。撤去干燥管,滴液漏斗缓慢逐滴滴入5 ml SOCl2,0℃继续反应30 min。撤掉冰浴,反应体系缓慢升温至室温后加热回流8 h,停止反应。除去过量SOCl2,残留物加入饱和NaHCO3溶液洗涤,氯仿萃取,有机层用水和饱和食盐水洗涤完毕后用Na2SO4干燥。减压蒸馏去氯仿,残余物经硅胶柱色谱(展开剂:石油醚/乙酸乙酯,v/v,2/ 1),得到目标产物无色油状液体,收率53%。IR(KBr):ν 3004(Ar-H),2961,2847,1681,1620,1442,1363,1163,815,659 cm-1;1H NMR(CDCl3,300 MHz):δ 7.65~7.70(t,1H,J=15 Hz,Py 4-H),7.40~7.42(d,2H,J=9 Hz,Py 3-H,5-H),3.89(s,4H,PyCH2N),3.52~3.57(t,8H,J=9 Hz,CH2OH),2.97~3.02(t,8H,J=12 Hz,NCH2);13C NMR(CDCl3,75 MHz)δ:158.2(pyridine 2-C),137.5(thienyl 4-C),121.0(thienyl 3-C),60.1(pyridine-CH2),56.3(NCH2),42.1(CH2Cl)ppm。
1.2.7 目标化合物2,6-双{[双(2-氯乙基)氨基]甲基}吡啶盐酸盐(8a)的合成 在100 ml圆底烧瓶中加入甲醇(5 ml)和化合物7(0.77 g,0.002 mol),室温搅拌下逐滴滴加4 mol/L的盐酸(2 ml),30℃继续反应24 h。TLC跟踪监测至原料点消失,停止反应。减压蒸馏除去过量盐酸,得到目标产物棕色固体,收率76%,m.p.212~215℃。1H NMR(CDCl3,300 MHz):δ 8.65~8.73(t,1H,J=15 Hz,Py 4-H),7.76~7.91(d,2H,J=6 Hz,Py 3-H,5-H),4.99(s,4H,PyCH2N),4.52~4.55(t,8H,J= 9 Hz,CH2OH),3.27~3.31(t,8H,J=12 Hz,NCH2)ppm。
1.2.8 目标化合物2,6-双{[双(2-氯乙基)氨基]甲基}吡啶硝酸盐(8b)的合成 在100 ml圆底烧瓶中加入乙酸乙酯(5 ml)、氯仿(5 ml)和化合物7(0.77 g,0.002 mol),室温搅拌下逐滴滴加4 mol/L的硝酸(1 ml),30℃继续反应24 h。TLC跟踪监测至原料点消失,停止反应。减压蒸馏除去过量盐酸,得到目标产物棕色固体,收率72%,m.p.218~221℃。1H NMR(CDCl3,300 MHz):δ 7.65~7.70(t,1H,J=15 Hz,Py 4-H),7.40~7.42(d,2H,J= 6 Hz,Py 3-H,5-H),3.89(s,4H,PyCH2N),3.52~3.57(t,8H,J=9 Hz,CH2OH),2.97~3.02(t,8H,J=12 Hz,NCH2)ppm;MS m/z:449[M+H-2HCl]+。
1.2.9 中间体2,6-吡啶二甲酰氯(9)的合成 氮气保护下,DMF(2 ml)慢慢加入到SOCl2(10 ml)中,室温下搅拌10 min后,加入2,6-吡啶二甲酸(3.34 g,0.02 mol),回流反应4 h。体系冷却至室温后,加入甲苯共沸除去过量SOCl2,残留物真空干燥后得到化合物9的粗品,不必纯化,直接投入下步反应。收率约61%,m.p.55~58℃。
1.2.10 中间体二(2-氯乙基)胺盐酸盐(10)的合成 在圆底烧瓶中加入氯仿(20 ml)和二乙醇胺(21 g,0.2 mol),冰浴冷却反应体系10 min,逐滴加入SOCl2(25 ml)。撤掉冰浴,室温下反应1 h后回流过夜反应。反应完毕后加入甲苯共沸除去过量SOCl2,残留物用丙酮重结晶,得到二(2-氯乙基)胺盐酸盐的白色固体,收率63%,m.p.216~218℃。
1.2.11 目标化合物N2,N2,N6,N6-四(2-氯乙基)吡啶-2,6-二甲酰胺(11)的合成 氮气保护、冰浴0~5℃条件下,在100 ml圆底烧瓶中加入化合物9(1.02 g,0.005 mol)、化合物10(1.42 g,0.01 mol)和15 ml干燥的THF,滴液漏斗缓慢逐滴滴入三乙胺(4 ml),继续反应30 min。让反应体系自然升到室温,继续反应30 h。停止反应,加入20 ml乙酸乙酯,硅藻土过滤,滤液旋蒸去溶剂,残余物经硅胶柱色谱(展开剂:石油醚/乙酸乙酯,v/v,1/1),得到目标产物棕油状液体,收率46%。IR(KBr)ν:2967,2852(CH3,CH2),1683(C=O),1257(C-N),1154 cm-1;1H NMR(CDCl3,300 MHz):δ 7.95~8.01(t,1H,J=20 Hz,Py 4-H),7.79~7.81(d,2H,J= 9 Hz,Py 3-H,5-H),3.89~3.85(t,J=12.0 Hz,8H,CH2Cl),3.68(bs,8H,CONCH2)ppm。
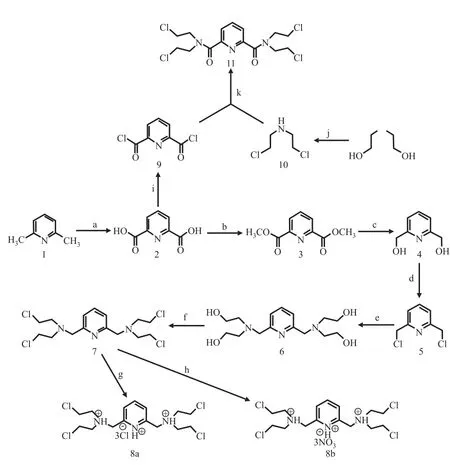
图1 吡啶氮芥类化合物的合成
2 结果
2.1 合成部分 在制备目标化合物的过程中,意外得到了中间体6的单晶,挑选出一个合适尺寸的完美单晶,并对其进行了X-射线衍射测试。
化合物6的椭球图见图2,其晶胞堆积见图3。单晶X射线衍射测试表明6的晶体结构属正交晶系P21212空间群,分子式为C15H27N3O4,晶胞参数为a=9.145(6)Â,b=10.716(7)Â,c=8.292(5)Â,V=812.6(9)Â3,Z=2。分子结构显示,化合物6的分子位于二重轴上,最小的不对称单元含半个分子,其中的一个N原子和一个C原子位于旋转轴上。吡啶环是氢键的受体,而两个羟基的O原子是氢键供体,形成了分子内的O—H···N和分子间的O—H···N,这样形成了一个闭合的氢键笼子。氢键数据显示,分子之间分子间的O—H·· ·N氢键相互作用有助于晶体的堆积形成。化合物6的更多的详细数据和精修参数,见辅助材料[15]。
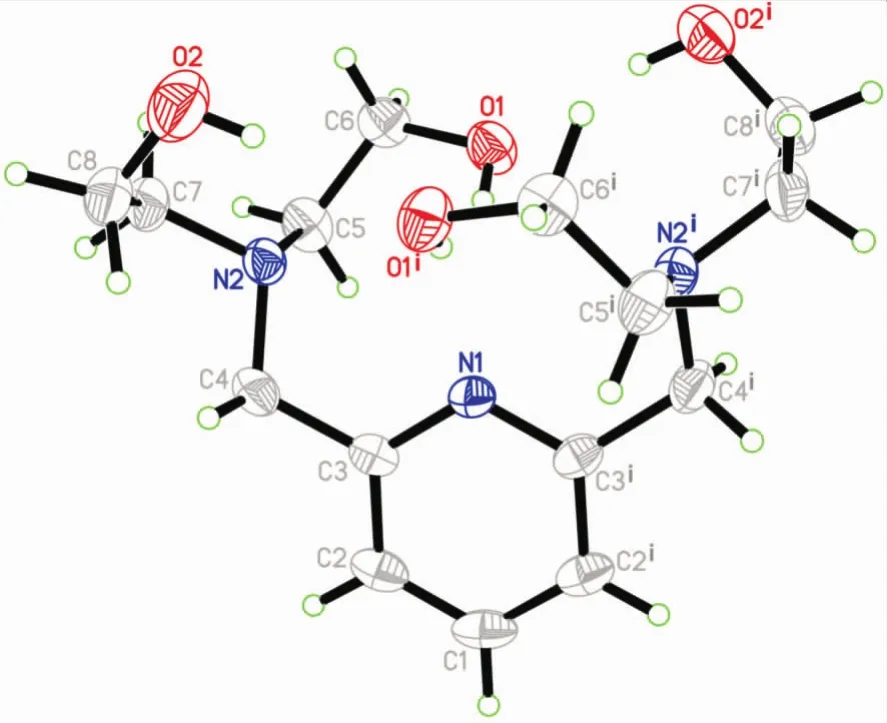
图2 化合物6的晶体结构
2.2 抗微生物活性部分 中间体及目标化合物的体外抗细菌、抗真菌活性数据见表1、表2。抗微生物活性测试所采用的药敏实验方法是美国国家实验室标准委员会(NCCLS)推荐的标准方法[16],选择氯霉素和氟康唑作为参考对照药物,并用最小抑制浓度(MIC,μg/ml)来表征化合物抗菌能力的大小。对照药物氯霉素是早期抗菌药,治疗浅表细菌感染的常用药物,而氟康唑是强效抗真菌药物,选择这两种药作为对照药具有一定的代表性。
体外抗细菌结果表明:目标化合物及中间体对所测细菌表现出不同程度的抑制活性,其中吡啶双氮芥化合物7、8a和8b对金黄色葡萄球菌的选择性抑制作用显著,MIC值为4 μg/ml,抑制活性与参照药物氯霉素相当。吡啶双酰胺氮芥化合物11对金黄色葡萄球菌的抑制能力很强(MIC=2 μg/ml),活性是氯霉素的2倍。但是这些化合物对所测的大肠杆菌、枯草杆菌、变形杆菌、铜绿假单胞菌和耐甲氧西林金黄色葡萄球菌的抑制效果普遍不佳。
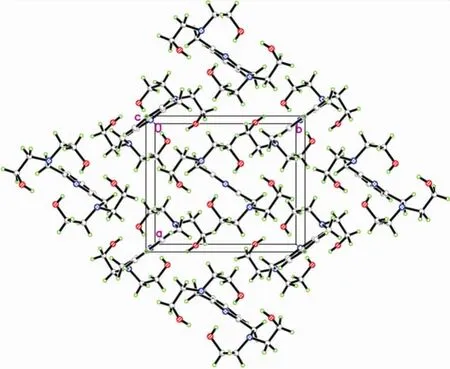
图3 化合物6沿A轴的晶胞堆积
体外抗真菌结果表明:目标化合物及中间体抗真菌的活性相比于其抗细菌活性要稍弱,部分化合物抑制活性不明显。吡啶双氮芥化合物7、8a、8b及11对白色念珠菌、假丝酵母菌有一定的抑制作用,MIC值为64~512 μg/ml,其中化合物11对白色念珠菌抑制活性最好,但弱于参照药物氟康唑。
3 讨论
在目标化合物的合成过程中,氯化是非常关键的步骤。化合物8和SOCl2反应,可以方便、迅速而高效地制备得到重要的中间体酰氯9,若想得到纯净的9可用石油醚进行重结晶。需注意的是,酰氯9不稳定,在空气中非常容易水解变回反应物2,6-吡啶二甲酸,所以无需精制可直接投入下步反应。
目标化合物7和11的合成,即f和k两步,对实验操作及溶剂的要求均很高,要求在氮气保护条件下且所用溶剂是新蒸无水溶剂,后处理过程也要迅速,否则产率过低或不能成功制备。步骤d、f、k三步使用无水非质子溶剂四氢呋喃,使反应活性和收率均比文献[17]值有所提高。目标化合物11的制备即k步需要控制反应的投料比,酰氯9和二(2-氯乙基)胺盐酸盐10的比例是1∶2,因为过多的二(2-氯乙基)胺盐酸盐在碱性条件下不仅会发生自身亲核取代,还会对11的四个氯乙基进行亲核取代。另外目标化合物7和11在常温、无氮气保护条件下快速变质,需要在低温、无水、无氧条件下保存,且不能长时间保存。
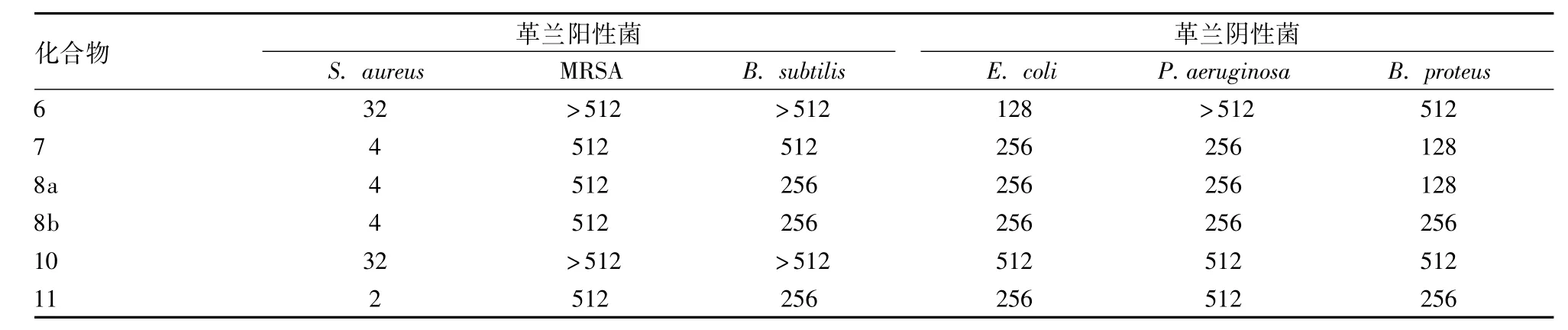
表1 目标化合物及中间体的抗细菌活性数据(MIC,μg/ml)
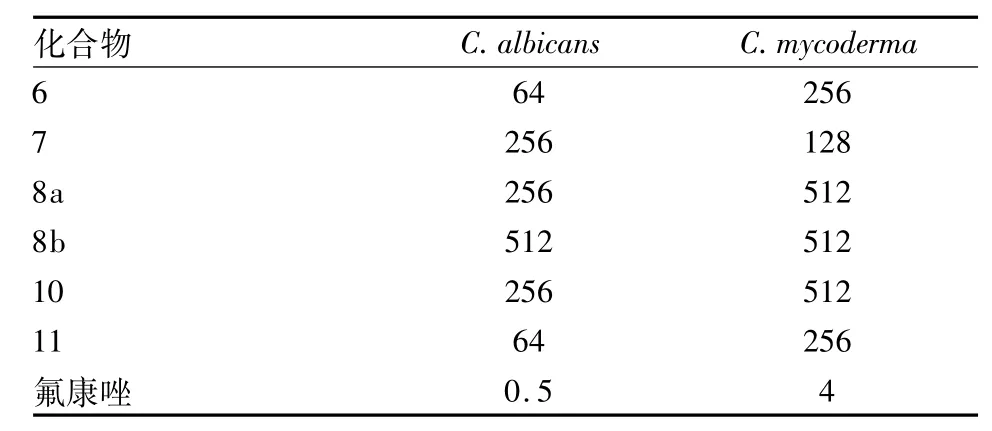
表2 目标化合物及中间体的抗真菌活性数据(MIC,μg/ml)
抗菌活性测试显示:目标化合物7和8a对变形杆菌的抑制作用优于其他测试化合物,MIC值为128 μg/ml。所测试的大部分化合物在一定程度上能够抑制被测微生物的生长,且目标化合物对金黄色葡萄球菌表现出很强的专一选择性抑制作用,活性与临床药物相当或优于临床药物,因此,此类化合物有望作为潜在的抗金葡菌先导化合物,值得进一步研究其构效关系。
[1] Bartzatt R,Cirillo S L,Cirillo J D,et al.Bifunctional constructs of aspirin and ibuprofen(non-steroidal anti-inflammatory drugs;NSAIDs)that express antibacterial and alkylation activities[J]. Biotechnol Appl Biochem,2003,37(Pt 3):273-82.
[2] 韩 亮,李正名,张 云,等.O-取代苯基-O-(2-硬脂酰胺基)乙基-N,N-二(2-氯乙基)磷酰胺的合成和生物活性[J].有机化学,2006,26(2):242-6.
[3] 庄雅云,周成合,王渝芳,等.氮芥类抗肿瘤药物研究进展[J].中国药学杂志,2008,43(17):1281-7.
[4] Zhao L M,Ma F Y,Jin H S,et al.Design and synthesis of novel hydroxyanthraquinone nitrogen mustard derivatives as potential anticancer agents via a bioisostere approach[J].Eur J Med Chem,2015,102:303-9.
[5] Li S,Wang X,He Y,et al.Design and synthesis of novel quinazoline nitrogen mustard derivatives as potential therapeutic agents for cancer[J].Eur J Med Chem,2013,67:293-301.
[6] 陈茹玉,王惠林,周 嘉.N′,N′-二-(2-氯乙基)-O-芳基-N3-芳基硫脲基磷酰胺酯的合成、结构及性质研究[J].高等学校化学学报,1995,16(8):1229-32.
[7] Rajski S R,Williams R M.DNA cross-linking agents as antitumor drugs[J].Chem Rev,1998,98(8):2723-96.
[8] 要晓丽,崔建丽,杨玉芬.吡啶类化合物的合成及应用研究进展[J].山西化工,2010,32(1):28-31.
[9] 徐 静.吡啶类化合物在医药领域的应用[J].中外医疗,2011,30(2):144.
[10]赵卫光,王建国,袁德凯,等.吡啶类农药研究趋势及新发现的含吡啶环的天然活性物质[J].农药,2002,41(7):8-11.
[11]张一宾.含吡啶环的新的农药及医药品种[J].研发前沿,2009,17(17):25-30.
[12]El-Zemity,Saad R.Antimicrobial activity of some 2-amino-5-subsituted pyridine derivatives[J].Arch Phytopathology Plant Protect,2011,44(4):381-9.
[13]Klimesova V,Svoboda M,Waisser K,et al.Antimicrobial activity of some 2-amino-5-subsituted pyridine derivatives[J].Farmaco,1999,54(10):666-72.
[14]Tang Y D,Zhang J Q,Zhang S L,et al.Synthesis and characterization of thiophene-derived amido bis-nitrogen mustard and its antimicrobial and anticancer activities[J].Chin J Chem,2012,30(1):1831-40.
[15]Cao S Y,Zhou C H.2,2′,2″,2‴-[Pyridine-2,6-diylbis(azanetriyl)]tetraethanol[J].Acta Crystallogr Sect E Struct Rep Online,2011,67(Pt 4):o1003.
[16]Özbek N,Katńrcioglu H,Karacan N,et al.Synthesis,characterization and antimicrobial activity of new aliphatic sulfonamide[J]. Bioorg Med Chem,2007,15(15):5105-9.
[17]Sprakel V S I,Elemans J A A W,Feiters M C,et al.Synthesis and characterization of PY2-and TPA-appended diphenylglycoluril receptors and their bis-CuI complexes[J].Eur J Org Chem,2006,10:2281-95.
Synthesis and antimicrobial evaluation of novel pyridine-derived nitrogen mustards
Cao Shouying1,Bai Changcun2,Tao Zhaolin1,et al
(1Dept of Chemistry,2Dept of Biochemistry&Molecular Biology,Bengbu Medical College,Bengbu 233030)
Objective A series of novel pyridine-derived nitrogen mustards were successfully synthesized via amulti-step sequence of oxidation,etherification,chlorination and N-alkylation.Methods The target compounds were confirmed by1H NMR,IR,MS spectra as well as elemental analyses.Results All these synthesized nitrogen mustards were evaluated for in vitro antimicrobial and fungi ctivities by two fold serial dilution technique.The antimicrobial activities indicated that most of the title compounds inhibited effectively all the tested stains in vitro and showed strongly selective inhibitory efficacy against Staphylococcus aureus with comparable or superior activity to the clinic drug.Among these compounds,pyridine-derived bis-alkyl nitrogen mustard 7 and its hydrochloride 8a and nitrateand 8b showed equivalent anti-S.aureus activities to chloramphenicol.Conclusion Pyridine-derived bisamide nitrogen mustard 11 exhibited better antibacterial activities against S.aureus than chloramphenicol,which were worthy of further investigation.A single crystal of the key intermediate 6 with suitable dimension and no crack was obtained and measured by X-ray diffraction.The structure belongs to orthorhombic system with space group P21212.
nitrogen mustard;pyridine;antibacterial;antifungal
O 622
A
1000-1492(2016)08-1145-06
时间:2016-6-22 14:44:58
http://www.cnki.net/kcms/detail/34.1065.R.20160622.1444.032.html
2016-05-04 接收
安徽省高等教育振兴计划(编号:2014zytz014);安徽省高等学校自然科学研究项目(编号:KJ2015B056by、KJ2015B126by);安徽省自然科学基金(编号:1308085QB24);安徽省生化药物工程技术研究中心开放课题(编号:BYEC1302)
蚌埠医学院1化学教研室、2生化与分子生物学教研室,蚌埠 233030
曹守莹,女,硕士,助教;
白长存,男,讲师,责任作者,E-mail:baichangcun@163. com
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
安徽化工(2022年1期)2022-02-15
汕头大学学报(自然科学版)(2020年4期)2020-12-14
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
天然产物研究与开发(2018年8期)2018-09-10
中成药(2017年5期)2017-06-13
中国资源综合利用(2016年7期)2016-02-03
股市动态分析(2015年12期)2015-09-10
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
