
基于高通量测序的云南松转录组分析
2016-11-10 12:51蔡年辉邓丽丽许玉兰王大玮何承忠段安安
植物研究 2016年1期
蔡年辉 邓丽丽 许玉兰 徐 杨 周 丽 王大玮 田 斌 何承忠 段安安
(1.西南林业大学云南省高校林木遗传改良与繁育重点实验室,昆明 650224; 2.西南林业大学国家林业局西南地区生物多样性保育重点实验室,昆明 650224; 3.西南林业大学西南山地森林保育与利用省部共建教育部重点实验室,昆明 650224)
基于高通量测序的云南松转录组分析
蔡年辉1,2,3邓丽丽1,2,3许玉兰1,2,3徐 杨1,2,3周 丽1,2,3王大玮1,2,3田 斌1,2,3何承忠1,2,3段安安1,2,3
(1.西南林业大学云南省高校林木遗传改良与繁育重点实验室,昆明 650224;2.西南林业大学国家林业局西南地区生物多样性保育重点实验室,昆明 650224;3.西南林业大学西南山地森林保育与利用省部共建教育部重点实验室,昆明 650224)
采用新一代高通量测序技术平台Illumina Hiseq 2 000对云南松转录组测序,得到的数据进行de novo组装,获得80 000条Unigenes,N50为1 881 nt、平均890 nt。与公共数据库进行比对,注释到NR、NT、Swiss-Prot数据库的Unigenes分别为43 434、46 415、29 418条。将Unigenes与COG数据库比对,有14 792条Unigenes成功注释,根据功能大致分成25类;与GO数据库比对,有26 743条Unigenes获得注释,按功能分为细胞组分、分子功能和生物过程3大类55亚类,其中参与的生物过程较多;以KEGG数据库参考,有25 873条Unigenes参与128条代谢途径分支,以代谢相关的通路较为集中,并找到与木质素合成关键酶的Unigenes。这些研究极大地扩充了云南松的基因资源,将有助于云南松基因的发掘与利用、分子标记的开发及其种质资源遗传改良的研究等。
云南松;转录组;Illumina高通量测序
云南松(PinusyunnanensisFranch.)是分布于我国亚热带山地暖性气候条件下的重要树种,是云贵高原的主要乡土树种和西南地区特有的森林植被类型[1~3],水平分布于东经96°~108°、北纬23°~30°[1,4~5],在云南省境内垂直分布于海拔710~3 320 m,大多分布于1 500~2 500 m[6]。具有适应性强、耐干旱瘠薄、喜光、木材用途广泛等特点,是分布区域内荒山造林的先锋树种和治理水土流失的重要树种,在分布区域林业生产和生态经济建设中占有举足轻重的地位,分别占云南省林分总面积和木材蓄积量的19.63%和14.28%[6~8]。目前云南松的研究以资源状况、遗传多样性、培育等方面报道较多,邓喜成等[6]通过对森林资源监测,比较云南松林的资源动态,表明云南松林可用资源数量仍在快速减少,材种结构低质化倾向加剧,迫切需要加强云南松林的抚育管理,科学开展森林经营活动;Xiao等[9]等提出云南松主要分布的区域,海拔2 500 m以下人类活动对资源的影响较大;Wu等[10~11]对云南松林的土壤因子分析,为云南松资源的保护与改良提供借鉴;Wang等[12]以云南松分布区的群体为对象,探讨云南松的遗传分化及其可能的成因,为开展云南松的基础研究提供指导,而转录组和基因组信息比较缺乏,对云南松分子标记开发也相对滞后[13]。
对于缺乏基因组信息的物种,采用转录组测序可获得大量的数据信息,有利于挖掘重要功能基因,是开展植物优良性状研究的重要手段[14~17]。随着高通量测序技术的不断发展,降低了测序的成本,同时获得丰富的数据,利于转录组的分析,有助于基因结构及其功能的研究,同时也为大量分子标记的开发奠定基础[18~22]。鉴于此,本研究采用Illumina HiSeq 2000高通量测序技术对云南松转录组测序分析,获得大量的数据,对其进行拼接与组装,建立云南松转录组数据库,结合生物信息学的方法对转录组数据库序列进行功能注释、功能分类和代谢通路分析,以期为云南松基因组水平上的研究奠定基础,同时也可为云南松EST-SSR分子标记的开发和功能基因挖掘提供数据。
1 材料与方法
1.1 试验材料
试验材料为永仁白马河云南松母树林采种育苗的5年生苗木,于2014年10月单株取样,采集当年生幼嫩的顶梢,然后迅速放入干冰中保存。
1.2 转录组测序
对采集的材料进行RNA提取,构建cDNA文库,采用Illumina Hiseq 2000平台测序。
1.3 序列组装
转录组测序后统计原始数据的数量及其长度。对原始数据经去除测序接头、重复冗余序列及低质量的序列数据等,获得clean reads,统计clean reads的数量、总长度、Q20、N%、GC%等,Q20表示过滤后质量不低于20的碱基的比例,N表示过滤后不确定的碱基的比例。采用Trinity软件(http://trinityrnaseq.github.io/)进行de novo组装,首先通过序列之间的overlap将序列延伸为Contig,再根据序列的双末端信息(paired-end reads),将Contig连接,得到该是样品的Unigene,分析Contig和Unigene的长度及其分布。
1.4 序列的注释、功能分类和生物学通路分析
获得的Unigene进行生物信息学的分析,包括功能注释及其功能分类。通过BLAST将拼接后的Unigene序列与蛋白数据库进行比对,以e-value<0.000 01为阀值获取注释,包括NCBI非冗余核酸数据库(Non-redundant protein database,NR)和Swiss-Prot蛋白质序列数据库(Swiss-Prot protein database,Swiss-Prot)。比对到NR数据库中,分析E-value、相似性及其物种的分布。根据NR注释信息,使用Blast2GO得到GO注释信息[23],GO即基因本体论数据库(Gene Ontology),是一个国际标准化的基因功能分类数据库,包括3个相对独立的类别,即参与的生物过程(Biological process)、构成的细胞组分(Cellular component)以及实现的分子功能(Molecular function),使用WEGO对所有的Unigene进行GO功能分类,从宏观上认识云南松基因功能分布特征[24]。对Unigene进行蛋白质直系同源数据库(Cluster of orthologous groups,COG)比对,对其可能的功能分类统计[25];同时也进行基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)代谢通路分析[26],进一步了解云南松的代谢途径、生物学功能以及基因间的相互作用等。此外,按NR、Swiss-Prot、KEGG和COG的优先顺序将Unigene与这些蛋白库进行blastx比对(e-value<0.000 01),将各Unigene比对到蛋白库的CDS(Coding DNA Sequence),未比对上的使用ESTScan预测出CDS。
2 结果与分析
2.1 云南松转录组测序及其de novo组装
测序共获得原始序列数据97 126 960条,对原始序列经去除测序接头、重复冗余、低质量等过滤处理,获得有效序列片段95 003 826条,其中,中间未知碱基序列的片段N为0,Q20高质量序列占94.9%,GC含量占总碱基数的45.67%。说明云南松转录组Illumina Hiseq 2000平台测序获得序列的数量及其质量均较高,为数据组装提供较好的原始序列数据,以满足后续的分析所用。
对测序的reads片段进行denovo组装,共获得122 115条序列重叠群(Contig),将这些Contig进一步组装,得到80 000条Unigenes,序列信息达71 215 401 nt,其中聚类(clusters)的Unigenes为25 226条,单独(singletons)的Unigenes为54 774条。
组装的序列长度也可以反映组装的质量,对contig的序列长度分析可知(图1:A),平均长度462 nt,N50为1 240 nt,其中200~1 000 nt长度的序列占87.89%,1 000~2 000 nt长度的序列占7.15%,2 000~3 000 nt的占3.14%,≥3 000 nt的序列占1.83%。对Unigene的长度进行统计(图1:B),平均长度为890 nt,N50为1 818 nt,有30%的序列长度大于1 000 nt,其中12%的大于2 000 nt。由此可以看出,通过转录组测序获得大量的序列,经组装后Unigenes的长度明显增加,平均长度增加近一倍。对于1 000 nt以上序列的分布也明显增加,所占比例由12%增加至30%,表明组装的效果较好,片段长度明显增加,可进一步开展后续分析。
2.2 云南松转录组Unigene的NR功能注释
将组装得到的80 000条Unigenes通过blast与NR库进行比对(evalue<0.000 01),43 434条Unigenes在NR数据库中找到相似序列,占总Unigenes的54.29%,结果如图2所示。
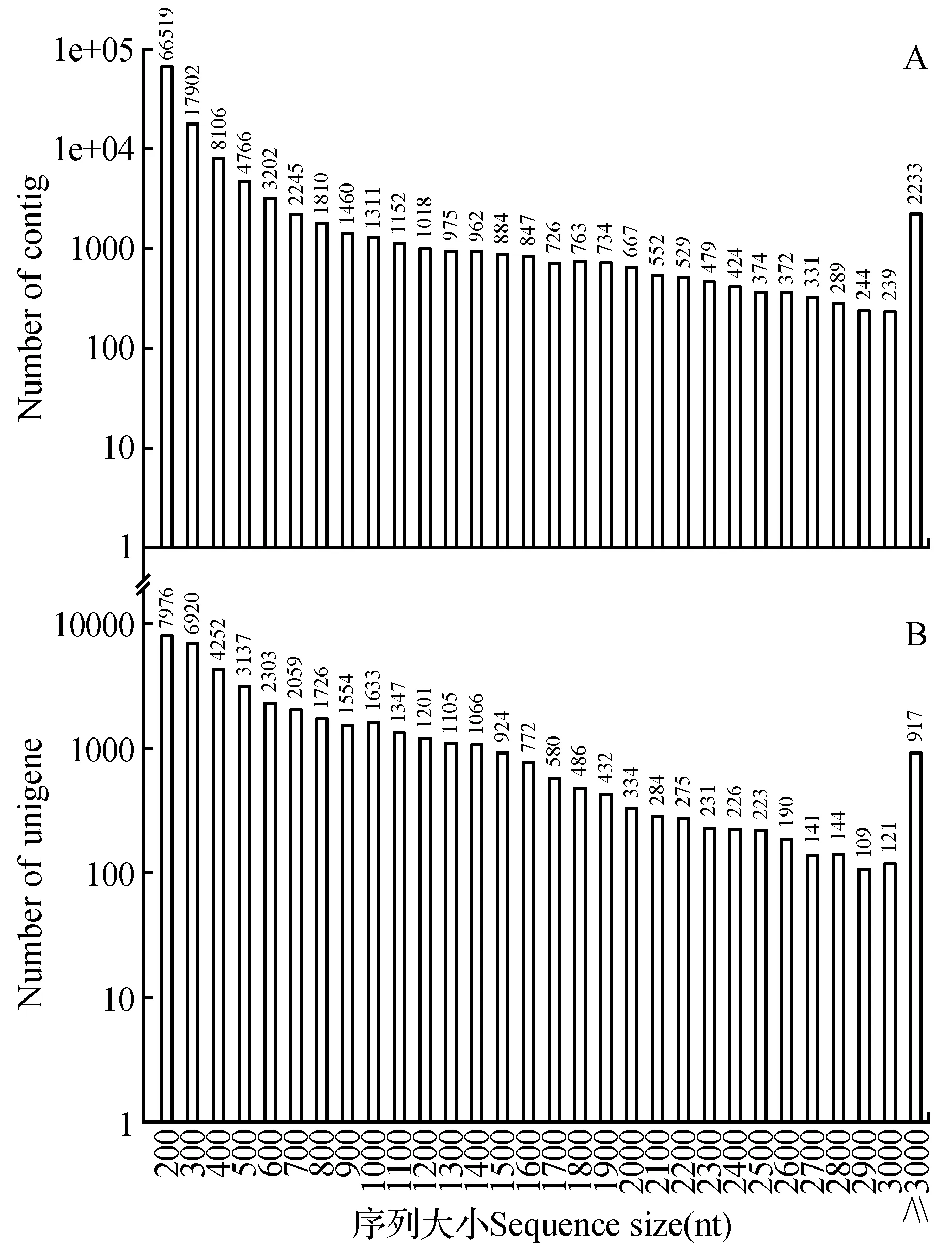
图1 云南松转录组组装序列长度分布 A. Contigs的长度分布;B. Unigenes的长度分布Fig.1 Length distribution of assembly contigs and Unigenes of transcriptome for P.yunnanensis A. Length distribution of assembly contigs; B. Length distribution of assembly unigenes
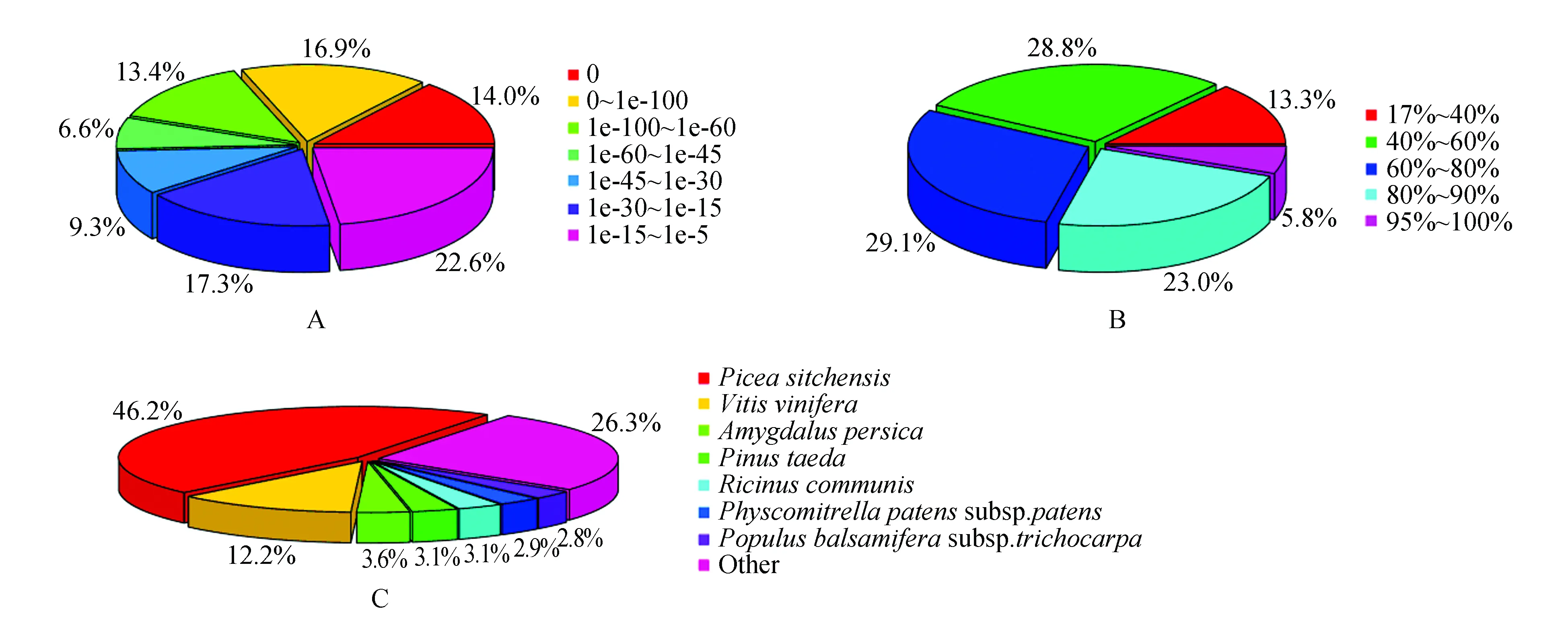
图2 云南松转录组Unigene的NR注释分类 A. NR注释的e-value分布;B. NR注释的相似度分布;C. NR注释的物种分布Fig.2 Category of NR annotation of transcriptome for unigenes of P.yunnanensis A. E-value distribution of NR annotation; B. Similarity distribution of NR annotation; C. Species distribution of NR annotation
E-value分布可以看出(图2:A),在注释的43 434条Unigenes中,有近一半(49.2%)的E值分布于e-45~e-5,36.9%的分布于e-100~e-45,在e=0的情况下占14%。从匹配序列的相似度分布可以看出(图2:B),有29.1%的序列的相似度在60%~80%,有28.8%的相似度>80%,42.1%的相似度<60%。从注释匹配的物种分布来看(图2:C),云南松43 434条Unigenes与其它物种已知基因具有不同程度的同源性,注释序列分布较多的5个物种分别为北美红杉(Piceasitchensis)、葡萄(Vitisvinifera)、桃(Amygdaluspersica)、火炬松(Pinustaeda)、蓖麻(Ricinuscommunis),分别占46.2%、12.2%、3.6%、3.1%和3.1%,其余近1/3的分布于其它400多个物种中。从E-value和相似度分布来看,云南松在NCBI的NR库中比对的匹配度较高,但由于缺乏云南松的转录组和基因组信息,部分Unigene在数据库中无法匹配到已知的基因中。
2.3 云南松转录组Unigene的COG注释及其分类
将获得的Unigenes与COG数据库进行比对,预测Unigenes可能的功能,根据比对结果对Unigenes的功能分类并统计(图3)。
结果表明,有14 792条Unigenes注释到COG库,占总Unigenes的18.49%,共获得29 002个COG功能注释信息,分布于25个功能分类,不同类别的基因表达丰度各不相同,以一般功能基因(General function prediction only)最多,占17.21%;其次是转录功能(Transcription),占9.04%;复制、重组和修饰(Replication,recombination and repair,8.59%),信号传递机制(Signal transduction mechanisms,7.51%),翻译后修饰、蛋白质折叠和分子伴侣(Posttranslational modification,protein turnover,chaperones,6.52%)以及碳水化合物转运和代谢(Carbohydrate transport and metabolism,5.94%)的功能基因也较丰富。而以核结构(Nuclear structure)和细胞外结构(Extracellular structures)的最少,分别仅有3个(0.01%)和26个(0.09%)。这些结果表明,云南松在转录、翻译和信号传递等基因表达丰度较高。此外,有2 089个功能注释信息未知,未确定其准确的生物学功能,占所有功能注释信息的7.20%。
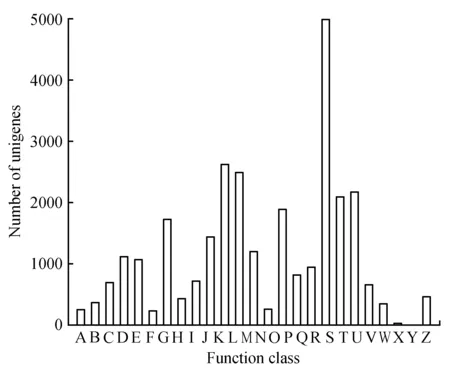
图3 云南松转录组Unigene的COG功能注释分布Fig.3 COG functional annotation distribution of unigenes of transcriptome for P.yunnanensis A.RNA processing and modification; B.Chromatin structure and dynamics; C.Energy production and conversion; D.Cell cycle control,cell division,chromosome partitioning; E.Amino acid transport and metabolism; F.Nucleotide transport and metabolism; G.Carbohydrate transoport and metabolism; H.Coenzyme transport and metabolism; I.Lipid transport and metabolism; J.Translation,ribosomal structure and biogenesis; K.Transcription; L.Replication,recombination and repair; M.Cell wall/membrane/envelope biogenesis; N.Cell motility; O.Posttranslational modification,protein turnover,chaperones; P.Inorganic ion transport and metabolism; Q.Secondary metabolites biosynthesis,transport and catabolism; R.General function prediction only; S.Function unknown; T.Signal transduction mechanisms; U.Intracellular trafficking,secretion,and vesicular transport; V.Defense mechanisms; W.Extracellular structures; Y.Nuclear structure; Z.Cytoskeleton
2.4 云南松转录组Unigene的GO注释及其分类
根据NR注释进一步进行GO功能注释,按照这些基因参与的生物过程、构成的细胞组分以及实现的分子功能进行分类统计,从宏观上了解云南松基因功能的分布特征,便于理解基因所代表的生物学意义(图4)。
分析可知(图4),共有26 743条Unigenes(占总Unigenes的33.43%)注释194 586个GO功能,对这些功能注释进行分类,生物过程、细胞组分和分子功能3大类分别注释94 703、70 538和29 345个基因,其中以生物过程注释的居多,占48.67%,其次是细胞组分,占36.25%,以分子功能较少(15.08%)。3个功能分类又可细分为55个功能亚类,分别包括22、17和16个亚类。在生物过程包含的22功能亚类中,以细胞过程(cellular process)、代谢过程(metabolic process)和单一有机体过程(single-organism process)较多,分别占该类型的15.93%、15.47%和11.25%,以运动(locomotion)最低,仅占生物过程全部的0.03%。细胞组分中以细胞(cell)和细胞组分(cell part)较多,均占该类别的25.52%,其次是细胞器(organelle),占该类别的20.00%,而以病毒体(virion)和病毒体组分(virion part)较低,两者均占该类别的0.003%。分子功能中以结合(binding)和催化活性(catalytic activity)居多,分别占该类型的47.74%和37.89%,而以通道调节活性(channel regulator activity)、翻译调节活性(translation regulator activity)、金属伴侣蛋白活性(metallochaperone activity)、蛋白标签(protein tag)较低,分别占该类别的0.003%、0.007%、0.014%和0.017%。这些结果显示了云南松新梢基因表达的总体情况,不难看出,以代谢活动相关的基因量较多,表明云南松代谢能力较强。
2.5云南松转录组Unigene的KEGG代谢通路分析
将Unigene比对到KEGG数据库,根据注释信息分析代谢通路,了解基因产物在细胞中的代谢途径及其基因产物的功能,结果表明共有25 873个Unigenes(32.34%)获得注释,对其可能参与或涉及的代谢通路统计分析,可将云南松Unigenes归为5个类别(Level 1)、20个亚类(Level 2)、128个代谢通路(Pathway)(图5)。
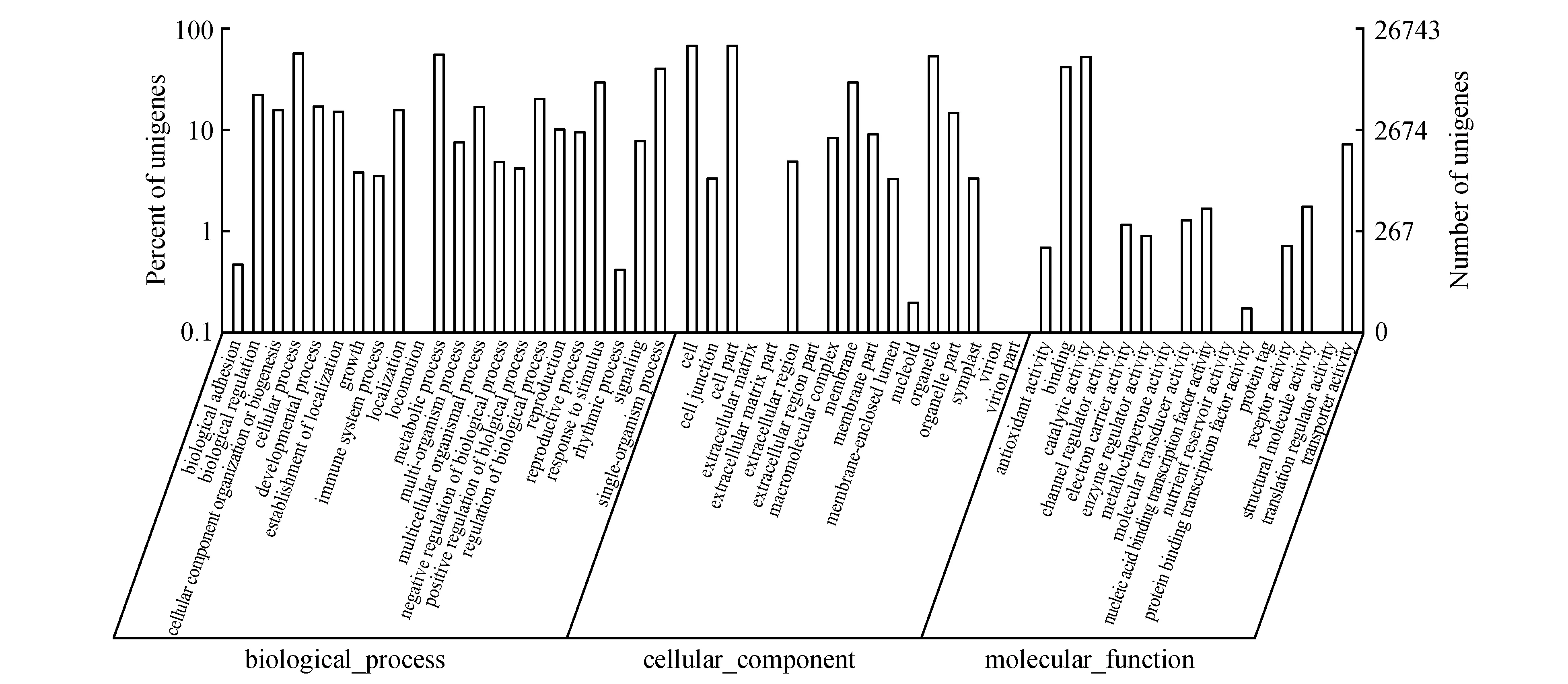
图4 云南松转录组Unigene的GO功能分类Fig.4 GO classification of unigenes of transcriptome for P.yunnanensis
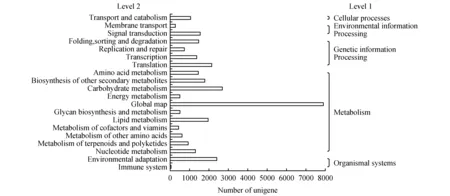
图5 云南松转录组Unigene的KEGG分类Fig.5 KEGG classification of unigenes of transcriptome for P.yunnanensis
由图5可以看出,5大类别中(Level 1),以代谢(Metabolism)相关的通路所占比例最多,为64.38%,其次为遗传信息处理(Genetic Information Processing)相关的通路,占18.43%,而以细胞过程(Cellular Processes)相关的通路最少,仅占3.43%,环境信息处理(Environmental Information Processing)和生物系统(Organismal Systems)相关的通路分别占5.85%和7.92%。进一步细分为亚类(Level 2),其中代谢相关的通路可细分为11亚类,包括氨基酸代谢(Amino acid metabolism)、其他次生物质代谢(Biosynthesis of other secondary metabolites)、碳水化合物代谢(Carbohydrate metabolism)、能量代谢(Energy metabolism)、全局整体映射(Global map)、糖生物合成和代谢(Glycan biosynthesis and metabolism)、脂类物质代谢(Lipid metabolism)、辅助因子和维生素代谢(Metabolism of cofactors and vitamins)、其它氨基酸代谢(Metabolism of other amino acids)、萜类化合物和聚酮化合物代谢(Metabolism of terpenoids and polyketides)和核苷酸代谢(Nucleotide metabolism)。而细胞过程仅包括运输和代谢(Transport and catabolism)1个亚类,其余遗传信息处理、环境信息处理和生物系统相关的通路分别包括4、2、2个亚类,共20个亚类。这些亚类中,所注释的Unigenes从高到低的依次为:全局整体映射(25.12%)、碳水化合物代谢(8.63%)、环境适应(Environmental adaptation,7.71%)、翻译(Translation,6.79%)、脂类物质代谢(6.33%)、其他次生物质代谢(5.77%)、信号传导(Signal transduction,4.96%)、折叠、分类和降解(Folding,sorting and degradation,4.78%)、氨基酸代谢(4.68%)、转录(Transcription,4.43%)、核苷酸代谢(Nucleotide metabolism,4.18%)、运输和代谢(3.43%)等,其中,以代谢相关的通路居多,表明云南松在这一时期具有较强的代谢活动。
5个Level 1、20个Level 2的Unigene定位到128个具体的KEGG代谢通路。按基因获得的注释量高低排序,将前20个代谢通路列于表1。从表1可以看出,以代谢途径(Metabolic pathway)的最多,占全部的16.42%,其次为次生代谢产物合成和植物病原互作(Plant-pathogen interaction),分别占8.70%和6.99%。
此外,为探讨与木质素合成相关Unigenes,本研究涉及到的苯丙类生物合成(Phenylpropanoid biosynthesis)的Unigenes有619条。进一步对该代谢通路进行分析,找到与木质素生物合成相关的酶(表2),包括苯丙氨酸氨裂解酶(phenylalanine ammonia-lyase,PAL)、4-香豆酰辅酶A连接酶(4-coumarate-CoA ligase,4CL)、莽草酸/奎宁酸羟基肉桂酰转移酶(shikimate O-hydroxycinnamoyltransferase,HCT)、咖啡酸辅酶A-O-甲基转移酶(caffeoyl-CoA O-methyltransferase,CCoAOMT)、肉桂酰辅酶A还原酶(cinnamoyl-CoA reductase,CCR)、阿魏酸-5-羟基化酶(ferulate-5-hydroxylase,F5H)、咖啡酸3-O-甲基转移酶(caffeic acid 3-O-methyltransferase,COMT)等。
表1云南松转录组Unigene的KEEGpathway注释
Table1TheKEEGpathwayannotationofunigenesoftranscriptomeforP.yunnanensis

代谢通路Pathway代谢通路编号PathwayID注释Unigene数Numnerofunigene比例Percentage(%)代谢途径Metabolicpathwayko01100515716.42次生代谢产物合成Biosynthesisofsecondarymetabolitesko0111027348.70植物病原互作Plant⁃pathogeninteractionko0462621966.99植物激素信号传导Planthormonesignaltransductionko0407513684.35剪接体Spliceosomeko030408512.71RNA转运RNAtransportko030138322.65嘧啶代谢Pyrimidinemetabolismko002406572.09嘌呤代谢Purinemetabolismko002306572.09淀粉和蔗糖代谢Starchandsucrosemetabolismko005006412.04苯丙生物合成Phenylpropanoidbiosynthesisko009406191.97内吞作用Endocytosisko041445271.68甘油磷脂代谢Glycerophospholipidmetabolismko005644901.56内质网蛋白加工Proteinprocessinginendoplasmicreticulumko041414741.51黄酮类化合物生物合成Flavonoidbiosynthesisko009414721.50RNA聚合酶RNApolymeraseko030204521.44mRNA监控途径mRNAsurveillancepathwayko030154351.38真核生物核糖体生物合成Ribosomebiogenesisineukaryotesko030084001.27RNA降解RNAdegradationko030183871.23泛素介导的蛋白降解途径Ubiquitinmediatedproteolysisko041203801.21核糖体Ribosomeko030103531.12
表2云南松转录组中参与木质素合成的酶
Table2TheenzymerelatedtoligninbiosynthesisoftranscriptomeforP.yunnanensis

EC号酶名称Enzymedefinition酶的缩写Enzymeabbr.编号EntryEC:4.3.1.24苯丙氨酸氨裂解酶Phenylalanineammonia⁃lyasePALK10775EC:6.2.1.124⁃香豆酰辅酶A连接酶4⁃coumarate⁃CoAligase4CLK01904EC:1.2.1.44肉桂酰辅酶A还原酶Cinnamoyl⁃CoAreductaseCCRK09753EC:2.1.1.68咖啡酸3⁃O⁃甲基转移酶Caffeicacid3⁃O⁃methyltransferaseCOMTK13066EC:2.1.1.104咖啡酸辅酶A⁃O⁃甲基转移酶Caffeoyl⁃CoAO⁃methyltransferaseCCoAOMTK00588EC:1.14.⁃.⁃阿魏酸⁃5⁃羟基化酶Ferulate⁃5⁃hydroxylaseF5HK09755EC:2.3.1.133莽草酸/奎宁酸羟基肉桂酰转移酶ShikimateO⁃hydroxycinnamoyltransferaseHCTK13065
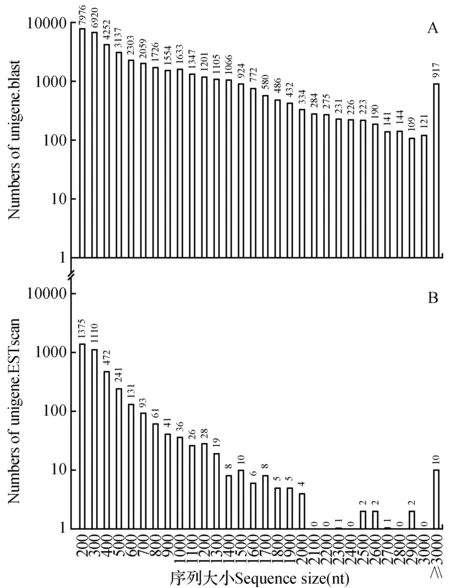
图6 云南松转录组Unigene的CDS序列长度分布Fig.6 CDS length distribution of transcriptome for P.yunnanensis
2.6 云南松转录组Unigene的CDS预测
按NR、Swiss-Prot、KEGG和COG的优先级顺序将Unigene序列与以上的蛋白库进行比对(evalue<0.000 01),不能比对的用ESTScan预测其编码区,结果有42 668条Unigenes能比对到蛋白库,另预测3 697条的CDS,其长度分布如图6所示。从图6A可以看出,比对的CDS序列长度1 000 nt以上的占26%,其中1 000~2 000 nt的占总数的19.33%,2 000~3 000 nt的占4.56%,3 000 nt以上的占2.15%。预测的CDS集中分布于200~1 000 nt,占96.29%,有极少数(0.27%)的序列在3 000 nt以上(图6B)。
3 讨论
研究采用Illumina高通量测序的数据量大,信息较多,是获得大量EST的有效途径,对于至今未开展基因组测序的物种而言,高通量测序是挖掘该物种生长发育过程中表达的重要基因可取途径,已应用于多种物种的基因研究,如辐射松(P.radiata)[14]、油松(P.tabuliformis)[15]、地中海白松(P.halepensisMill.)[16]、高山松(P.densata)[17]等。本研究通过云南松转录组测序,经数据组装后获得80 000条Unigenes,与庞大的松树全基因组相比[27],本研究获得的信息量大,可为后续的基因功能分析、基因克隆、分子标记的开发等方面奠定基础。通过序列分析,组装后获得的Unigene序列长度平均为890 nt,与其它松树如马尾松[28~29]、油松[15]、地中海白松[16]等相比,本研究云南松转录组序列拼接平均长度较好,与此同时,组装后的N50达到1 818 nt,序列较长。总体来看,从序列的数量、序列的Q20、序列的长度分布、N值等方面比较,获得的序列数量和质量都比较高,有利于后续的研究分析。测序拼接后的序列clusters有25 226条、singletons有54 774条,singleton的序列比较多,可能是拼接时严格的参数选择或测序时低表达基因的出现等,其中大量的singleton也获得注释,说明这些singleton也代表转录组信息,为高质量有用的reads,这在柳树(Salixspp.)的研究中也提到[30]。
采用生物信息学分析方法,对云南松转录组的序列进行功能注释及其功能分类。通过NR数据库的比对,有43 434条序列比对上,占总Unigene的54.29%。通过COG数据库比对,获得了大量多方面的表达基因,这些基因反映了云南松在一定时期的表达情况。按其功能可把14 792条序列包含的基因分为25类,其中以一般功能、转录较为丰富,而有7.2%(2 089条)的Unigenes功能未知,难以确定其生物功能,可能是注释信息不完善造成的,在很多林木转录组分析中也出现同样的情况,如泡桐(Paulownia)[31]、辽东栎(Ouercusliaotungensis)[32]、锥栗(Castaneahenryi)[33]、柳树[30]等。转录组中的Unigenes根据GO功能分为生物过程、细胞组分和分子功能3大类55个亚类,以分布于细胞过程、代谢过程、细胞、细胞组分、细胞器、结合和催化活性等方面较为集中;根据KEGG代谢通路分析,共有25 873个Unigenes(32.34%)注释到对应的128个代谢通路中,其中以代谢相关通路的占多数,表明云南松具有比较丰富的代谢活动。通过注释的信息量来看,云南松基因含量丰富,为其适应性强提供了分子生物学方面的解释与支持。尽管这些Unigenes并不覆盖云南松整个蛋白编码区,但这些数据也提供较为丰富的表达信息,为今后云南松功能基因的挖掘、利用、分子标记的开发奠定基础。总的未注释Unigenes有27 825条,占总Unigene的34.78%,这些序列可能是因为本身为非编码的RNA序列,或者是因长度不足未包含蛋白质功能域的信息,也有可能是数据库中的基因信息不足或云南松特有的新基因未能匹配上[29]。
4 结论
通过转录组测序,获得的Unigenes与公共数据库进行比对,有65.22%的Unigenes获得注释信息,其中COG分析将其分为25个不同功能分类,以一般功能基因和转录功能的较多;GO分析得到55个不同功能分类,以生物学过程占多数,各亚类中以细胞过程、代谢过程等类别的基因含量丰富,表明云南松具有强的代谢活力及其代谢机制;KEGG的128个代谢通路,以涉及代谢途径和次生代谢产物合成较为丰富,并找到涉及到木质素合成的关键酶多个,有助于将来开展云南松材质的研究。研究结果揭示了云南松基因丰富,并分析基因的分布特征,确定基因代谢通路的分布,为云南松较强的适应性提供了转录组水平上的支持,也为后续云南松基因组学、功能基因、分子标记的开发等方面的研究奠定基础。
1.金振洲,彭鉴.云南松[M].昆明:云南科技出版社,2004:1-66.
2.中国科学院昆明植物研究所编著.云南植物志(第4卷):种子植物[M].北京:科学出版社,1986:54-57.
3.中国科学院中国植物志编辑委员会.中国植物志:第7卷[M].北京:科学出版社,1978:122-282.
4.陈飞,王健敏,孙宝刚,等.云南松的地理分布与气候关系[J].林业科学研究,2012,25(2):163-168.
5.陈飞,王健敏,陈晓鸣,等.基于Kira指标的云南松气候适宜性分析[J].林业科学研究,2012,25(5):576-581.
6.邓喜庆,皇宝林,温庆忠,等.云南松林资源动态研究[J].自然资源学报,2014,29(8):1411-1419.
7.邓喜庆,皇宝林,温庆忠,等.云南松林在云南的分布研究[J].云南大学学报:自然科学版,2013,35(6):843-848.
8.Zhang L,Xu W H,Ouyang Z Y,et al.Determination of priority nature conservation areas and human disturbances in the Yangtze River Basin,China[J].Journal for Nature Conservation,2014,22(4):326-336.
9.Xiao X Y,Haberle S G,Shen J,et al.Latest Pleistocene and Holocene vegetation and climate history inferred from an alpine lacustrine record,northwestern Yunnan Province,southwestern China[J].Quaternary Science Reviews,2014,86(2):35-48.
10.Wu J X,Wang Y X,Chen Q B,et al.Soil improvement of Pinus yunnanensis forest at different age in Central Yunnan Plateau[J].Advanced Materials Research,2014,864-867:2565-2568.
11.Lin Y M,Cui P,Ge Y G,et al.The succession characteristics of soil erosion during different vegetation succession stages in dry-hot river valley of Jinsha River,upper reaches of Yangtze River[J].Ecological Engineering,2014,62(1):13-26.
12.Wang B S,Mao J F,Zhao W,et al.Impact of Geography and Climate on the Genetic Differentiation of the Subtropical PinePinusyunnanensis[J].PLoS ONE,2013,8(6):e67345.
13.Xu Y L,Zhang R L,Tian B,et al.Development of novel microsatellite markers forPinusyunnanensisand their cross amplification in congeneric species[J].Conservation Genetics Resources,2013,5(4):1113-1114.
14.Li X,Yang X,Wu H X.Transcriptome profiling of radiata pine branches reveals new insights into reaction wood formation with implications in plant gravitropism[J].BMC Genomics,2013,14(22):547-554.
15.Niu S H,Li Z X,Yuan H W,et al.Transcriptome characterisation ofPinustabuliformisand evolution of genes in thePinusphylogeny[J].BMC Genomics,2013,14(1):202-207.
16.Pinosio S,González-Martínez S C,Bagnoli F,et al.First insights into the transcriptome and development of new genomic tools of a widespread circum-mediterranean tree species,PinushalepensisMill.[J].Molecular Ecology Resources,2014,14(4):846-856.
17.Wan L C,Zhang H,Lu S,et al.Transcriptome-wide identification and characterization of mirnas fromPinusdensata[J].BMC Genomics,2012,13(1):132-142.
18.Fang P,Niu S,Yuan H,et al.Development and characterization of 25 EST-SSR markers inPinussylvestrisvar.mongolica(Pinaceae)[J].Applications in Plant Sciences,2014,2(1):1300057.
19.Feng Y H,Yang Z Q,Wang J,et al.Development and characterization of SSR markers fromPinusmassonianaand their transferability toP.elliottii,P.caribaeaandP.yunnanensis[J].Genetics & Molecular Research Gmr,2014,13(1):1508-1513.
20.Iwaizumi M G,Tsuda Y,Ohtani M,et al.Recent distribution changes affect geographic clines in genetic diversity and structure ofPinusdensifloranatural populations in Japan[J].Forest Ecology & Management,2013,304(4):407-416.
21.Lesser M R,Parchman T L,Buerkle C.Cross-species transferability of SSR loci developed from transciptome sequencing in lodgepole pine[J].Molecular Ecology Resources,2012,12(3):448-455.
22.Liu J J,Hammett C.Development of novel polymorphic microsatellite markers by technology of next generation sequencing in western white pine[J].Conservation Genetics Resources,2014,6(3):647-648.
23.Conesa ,Götz S,García-Gómez J M,et al.Blast2GO:A universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21(18):3674-3676.
24.Ye J,Fang L,Zheng H,et al.WEGO:a web tool for plotting GO annotations[J].Nucleic Acids Research,2006,34(2):293-297.
25.Tatusov R L,Galperln M Y,Natale D A,et al.The COG database:A tool for genome-scale analysis of protein functions and evolution[J].Nucleic Acids Research,2000,28(1):33-36.
26.Kanehisa M,Goto S,Kawashima S,et al.The KEGG resource for deciphering the genome[J].Nucleic Acids Research,2004,32(22):277-280.
27.Zimin A,Stevens K A,Crepeau M W,et al.Sequencing and assembly of the 22-gb loblolly pine genome[J].Genetics,2014,196(3):875-890.
28.王晓峰,何怀卫龙,蔡卫佳,等.马尾松转录组测序和分析[J].分子植物育种,2013,11(3):385-392.
29.Bai T D,Xu L,Xu M,et al.Characterization of masson pine(PinusmassonianaLamb.) microsatellite DNA by 454 genome shotgun sequencing[J].Tree Genetics & Genomes,2014,10(2):429-437.
30.郑纪伟.柳树转录组高通量测序及SSR标记开发研究[D].南京:南京林业大学,2013.
31.邓敏捷,董焱鹏,赵振利,等.基于Illumina高通量测序的泡桐转录组研究[J].林业科学,2013,49(6):30-36.
32.刘玉林,李伟,张志翔.基于高通量测序的辽东栎转录组学研究[J].生物技术通报,2014(7):119-124.
33.张琳,范晓明,林青,等.锥栗种仁转录组及淀粉和蔗糖代谢相关酶基因的表达分析[J].植物遗传资源学报,2015,16(3):603-612.
TranscriptomeAnalysisforPinusyunnanensisBasedonHighThroughputSequencing
CAI Nian-Hui1,2,3DENG Li-Li1,2,3XU Yu-Lan1,2,3XU Yang1,2,3ZHOU Li1,2,3WANG Da-Wei1,2,3TIAN Bin1,2,3HE Cheng-Zhong1,2,3DUAN An-An1,2,3
(1.Southwest Forestry University,Key Laboratory for Forest Genetic and Tree Improvement & Propagation in Universities of Yunnan Province,Kunming 650224;2.Key Laboratory of Biodiversity Conservation in Southwest China,State Forestry Administration,Southwest Forestry University,Kunming 650204;3.Key laboratory for Forest Resources Conservation and Use in the Southwest Mountains of China,Ministry of Education,Southwest Forestry University,Kunming 650224)
The transcriptome ofPinusyunnanensiswas sequenced by using Illumina Hiseq 2 000. In total 80 000 Unigene with an average length of 890 nt and N50 of 1 881 nt were obtained by de novo assembly. Of the Unigene, 43 434, 46 415 and 29 418 Unigenes had significant similarity with known data bank in NR, NT and Swiss-Prot, respectively. 14 792 Unigenes were annotated in clusters of orthologous groups of proteins(COG) and assigned to 25 clusters. 26 743 Unigenes were annotated in gene ontology(GO) and grouped into biological processes, cellular components and molecular function three functional categories, 55 sub-categories. The biological processes were most commonly existed. A total of 25 873 Unigenes were divided into 128 Kyoto Encyclopedia of Genes and Genomes(KEGG) pathways whose functions focused on metabolism. We found some Unigenes related to lignin biosynthesis. The sequence data forP.yunnanensiswiill be helpful for the gene discovery and utilization, molecular marker development and genetic improvement in the further research.
Pinusyunnanensis;transcriptome;Illumina high throughput sequencing technology
国家自然科学基金项目(31360189)和(31260191)
蔡年辉(1975—),男,硕士,讲师,主要从事森林培育研究。
2015-06-12
S791.257
A
10.7525/j.issn.1673-5102.2016.01.011
猜你喜欢
草业学报(2022年3期)2022-03-26
传染病信息(2021年6期)2021-02-12
林产工业(2020年8期)2020-08-31
科海故事博览·下旬刊(2019年6期)2019-04-16
乡村科技(2019年9期)2019-02-22
山西农业科学(2019年10期)2019-02-12
中国生殖健康(2018年4期)2018-11-06
中国实验诊断学(2017年5期)2017-06-05
广西林业科学(2016年3期)2016-03-16
化学工业与工程(2015年1期)2015-02-10
