
C2N纳米带的磁性和电子特性
2016-10-21 06:40顾利萍于海林
常熟理工学院学报 2016年4期
顾利萍,于海林
(常熟理工学院a.物理与电子工程学院;b.江苏省新型功能材料重点建设实验室,江苏常熟215500)
C2N纳米带的磁性和电子特性
顾利萍a,b,于海林a,b
(常熟理工学院a.物理与电子工程学院;b.江苏省新型功能材料重点建设实验室,江苏常熟215500)
采用基于密度泛函理论的第一性原理计算对C2N纳米带的磁性和电子特性进行了研究.对宽度最小的zigzag类型的纳米带,考虑了边缘原子的无H饱和(ZCNNR)、部分H饱和(ZCNNR-H)和完全饱和(ZCNNR-2H)3种情况.通过对其磁性和电子特性的分析发现,ZCNNR和ZCNNR-H的单胞磁矩分别为12 μB和4 μB,并表现出自旋半导体的特性,而ZCNNR-2H表现出非磁的半导体性质.进一步通过对自旋电荷密度的分析发现,其磁性和电子特性与边缘的N原子的H饱和密切相关.
第一性原理计算;磁性;能带;态密度
1 引言
二维材料由于其具有非常奇异的电子特性、磁性和力学特性,因而近年来受到广泛的关注[1-3].最早发现的二维材料为Graphene,即石墨的单原子层结构,其具有超高的电导率和热导率.然而,由于Graphene在费米面没有带隙存在,导致其在场效应晶体管和光电器件中的应用受到限制.因此,探索新的具有适当带隙的二维半导体材料是一件非常有意义的工作.最近,这一方面的研究工作取得了重要突破.一种全新的二维材料,多孔的C2N(C2N-h2D)成功制备出来[4].C2N-h2D有严格的二维晶体结构,并且具有1.96 eV的直接带隙.更有趣的是基于C2N-h2D制备的场效应晶体管,其开/关比高达107,这意味着C2N-h2D在电子器件和光电子器件中具有重大的应用价值.
自从C2N-h2D成功制备以来,吸引了许多研究工作的兴趣.Xu[5]等对C2N-h2D的气体分子的透过性进行了研究,发现H2分子的透过性要远远高于大气中的其他气体分子,表明其在氢分离方面有重要的潜在应用.Yang[6]等对几个原子层厚的C2N-h2D的电子特性进行了研究.Sahin[7]等研究了C2N-h2D的结构、声子和热学特性.Kang[8]等研究了由graphene和C2N-h2D组成的异质结的电子结构特性.Yang[9]等研究了应变对C2N-h2D的电子结构和光学特性的影响.Senger[10]等对二维多孔晶体C2X(X=N,P or As)的结构、电子特性和力学性能进行了研究.最近,Zhao等的结果表明C2N-h2D在轻元素的同位素,如3He/4He等的分离上有重要的研究价值.
从应用的角度来看,理解和掌握C2N-h2D纳米带的磁性和电子特性是非常必要的,因为纳米带的形状和边缘原子悬挂健的饱和程度对其性能有重要的影响.然而,到目前为止还没有关于C2N-h2D纳米带的磁性和电子特性的研究报道.出于对C2N-h2D纳米带的物理特性理解的需要,以及其在工程技术中的潜在应用,在本工作中我们利用第一性原理计算对zigzag类型的C2N-h2D纳米带(ZCNNRs)的磁性和电子特性进行计算.我们发现ZCNNRs的磁性和电子特性与其边缘原子悬挂健的饱和程度密切相关.同时也研究了外加电场对ZCNNRs带隙的调控.
2 计算模型
C2N-h2D的晶体结构如图1所示,为二维的多孔结构,有严格的二维周期性.最小宽度的ZCNNR如图1(b)中的虚线所示.考虑到ZCNNRs构建的时候,其边缘的C-N健会断裂,导致其边缘原子会产生悬挂健,因而在本工作中也对边缘原子用H饱和的结构进行了研究.对于ZCNNRs,其边缘有两种类型的N原子,一种是和一个C原子成键(N1),另一种是和两个C原子成键(N2),如图2(a)所示.N1原子有两个悬挂键,因此,本工作中考虑的两种H饱和方式,如图2(b)和(c)所示.
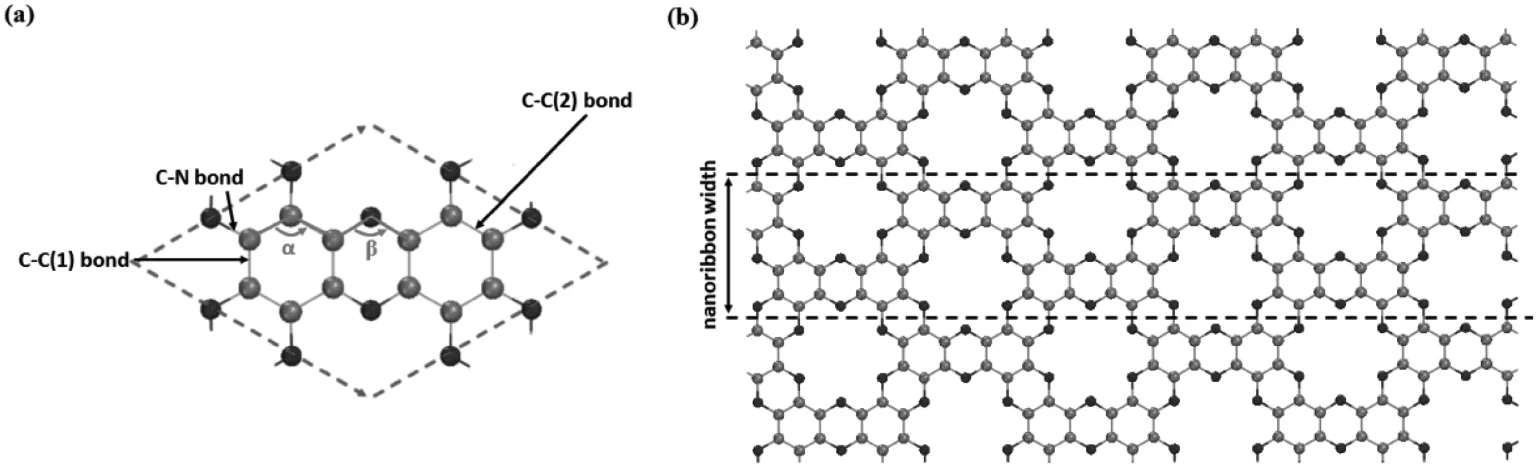
图1 C2N-h2D的结构示意图(a)C2N-h2D的原胞结构;(b)C2N-h2D的二维周期性结构,虚线所示为所选取的纳米带的示意图
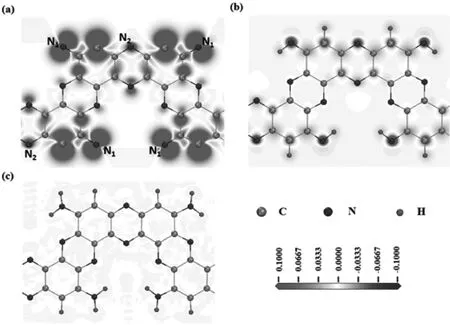
图2 最小宽度的ZCNNRs结构示意图(a)没有H饱和的ZCNNR,(b)N1有一个H饱和的ZCNNR-H,(c)N1有两个H饱和的ZCNNR-2H
3 计算方法
本工作的所有计算均采用基于第一性原理的VASP软件包进行.采用的交换-关联泛函为广义梯度近似,所选用的势文件为Perdew-Burke-Ernzerhof势.平面波的阶段能选取500 eV,布里渊区K点的采样使用Monkhorst-Pack方法,并取为1×1× 15.计算中所使用超胞的真空层厚度为20 Å,足以避免由于周期性所带来的相邻单胞之间的相互作用.在我们的计算中电子步的收敛标准设置为10-6eV,离子收敛标准设置为0.005 eV/Å.
4 结果分析
优化以后的ZCNNRs结构如图2所示,其边缘的C-N键、C-C(1),C-C(2)键和C-C-C(α)以及C-NC(β)角(对应图1(a)中的值)列于表1中.从表1中我们发现这些纳米带边缘原子形成的键和角相对二维C2N-h2D片的值变化都比较小,变化最大的为ZCNNR,其键长和键角的相对最大变化值为0.061 Å和7.8°.我们的计算结果表明这些纳米带具有很好的稳定性.
C2N-h2D的响应值和纳米带单胞的磁矩也列在表1中.

表1 优化以后的C-N、C-C(1)、C-C(2)键的键长及C-C-C(α)和C-N-C(β)角
计算得到的这些纳米带的磁矩也列于表1中.从表1中我们发现其磁矩和边缘原子的饱和程度密切相关.对于ZCNNR结构,其边缘有4个N1原子核,4个未饱和C原子,其未成对的电子为12个,对应其单胞磁矩为12 μB.对于ZCNNR-H结构,其边缘的4个C原子被H原子饱和,同时4个N1原子被4个H原子饱和,这样只有4个未成对电子,对应的磁矩恰好为4 μB.而ZCNNR-2H结构,边缘所有的未成对电子都被H原子饱和,其对应的磁矩恰好为0.为了进一步理解纳米带的磁性特性,我们计算了自旋电荷密度的分布,如图2所示.从图中可以看出,纳米带的磁矩基本上完全由边缘未饱和原子决定.
为了进一步理解这些纳米带的电子结构特性,我们计算了其能带和相应的态密度,如图3-图5所示.从图3和图4可以看出,ZCNNR和ZCNNR-H纳米带均表现出自旋半导体特性,其带隙分为0.46 eV和0.14 eV.而ZCNNR-2H自旋向上和向下的能带完全重合,表现出经典的半导体特性,其带隙为1.38 eV.

图3 ZCNNR纳米带的能带和相应的态密度

图4 ZCNN-H纳米带的能带和相应的态密度

图5 ZCNN-2H纳米带的能带和相应的态密度
为了探讨决定其电子结构特性的因素,我们对这些纳米带价带顶和导带底的能带所对应的电荷密度进行了计算,结果如图6-图8所示.从图6-图8我们可以发现其导带顶和价带低均由边缘的原子决定.因此,我们对边缘原子的局域态密度进行了计算,发现纳米带边缘的N1和C原子的px轨道对其价带顶和导带底的贡献最大.考虑到纳米带为一平面结构,x方向垂直于纳米带表明,C原子和N之间以sp2方式成键,这样导致px轨道对成键贡献最小.因而费米面附近的电子性质由px轨道决定.
5 结论
在本工作中,我们利用第一性原理计算对zigzag类型的C2N纳米带的磁性和电子特性进行了研究.计算结果表明C2N纳米带的磁性和纳米带边缘原子的H饱和度密切相关.对于没有H饱和的纳米带,其单胞磁矩为12 μB,对于部分H饱和的纳米带,其单胞磁矩为4 μB,对于全部H饱和的纳米带,其单胞磁矩为0.从纳米带的能带结构特性来看,未饱和和部分饱和的纳米带为自旋半导体特性,其带隙为0.46 eV和0.14 eV,而全部饱和的纳米带,为经典的半导体性质,其带隙为1.38 eV.

图6 (a)ZCNNR纳米带导带底和价带顶所对应的电荷密度,(c)-(d)为边缘C和N1、N2原子的态密度

图7 (a)ZCNNR-H纳米带导带底和价带顶对应的电荷密度,(c)-(d)为边缘C和N1、N2原子的态密度

图8 (a)ZCNNR-2H纳米带导带底和价带顶对应的电荷密度,(c)-(d)为边缘C和N1、N2原子的态密度
[1]ZHE W,DONG-KEUN K,HUA C,et al.MacDonald&Alberto F.Morpurgo,Strong interface-inducedspin-orbit interaction in graphene on WS2[J].NATURE COMMUNICATIONS,2015,6:8339.
[2]AVINASH P,NAYAK,ZHEN Y,et al.Pressure-Modulated Conductivity,Carrier Density,and Mobility of Multilayered Tungsten Disulfide[J].ACS NANO,2015,9(9):9117-9123.
[3]DARWIN B P,SHI-HSIN L,JER-LAI K.A first-principles examination of conducting monolayer 1T`-MX2(M=Mo,W;X=S,Se,Te):promising catalysts for hydrogen evolution reaction and its enhancement bystrain[J].Phys Chem Chem Phys,2015,17,21702.
[4]MAHMOOD J,M JUNG E K LEE,et al.Nitrogenated holey two-dimensionalstructures[J].NATURE COMMUNICATIONS,2015,6:6486.
[5]XU B,XIANG H,WEI Q,et al.Two-dimensional graphene-like C2N:an experimentally available porous membrane for hydrogen purification[J].Phys Chem Chem Phys,2015,17:15115.
[6]ZHANG R Q,LI B,YANG J L.Effects ofstacking order,layer number and external electric field on electronicstructures of fewlayer C2N-h2D[J].NANOSCALE,2015,7(33):14062-14070.
[7]SAHIN H.Structural and phononic characteristics of nitrogenated holey graphene[J].PHYSICAL REVIEW B,2015,92(8): 085421.
[8]KANG J,HORZUMs.Heterostructures of graphene and nitrogenated holey graphene:Moir'e pattern and Dirac ring[J].PHYSICAL REVIEW B,2015,92:195419.
[9]GUANs,CHENG Y C,LIU C,et al.Effects ofstrain on electronic and optic properties of holey two-dimensional C2N crystals[J]. APPLIED PHYSICS LETTERS,2015,107:231904.
[10]YAGMURCUKARDES M,HORZUMs,TORUN E,et al.phosphorated and arsenicated monolayer holey graphenes[J].Phys Chem Chem Phys,2016,18:3144-3150.
Magnetic and Electronic Properties of C2N nanoribbons
GU Lipinga,b,YU Hailina,b
(a.School of Physics and Electronic Engineering;b.Jiangsu Laboratory of Advanced Functional Materials,Changshu Institute of Technology,Changshu 215500,China)
The magnetic and electronic properties of C2N have beenstudied using first-principles calculations based on density functional theory.For the zigzag nanoribbons with thesmallest band width,three kinds of nanoribbons,i.e.without H passivation(ZCNNR),with one H passivation(ZCNNR-H)and with two H passivation(ZCNNR-2H)for edge N atom have been considered.For ZCNNR and ZCNNR-H,the magnetic moments are 12μB and 4μB respectively,and have presented thespinsemiconductor properties,while for ZCNNR-2H,itshows non magnetic property.Based on the analysis ofspin electronic density,the magnetic and electronic properties of these nanoribbons are closely related to the passivation of edge N atoms.
first-principles;magnetic;bandstructure;density ofstates
O482.6
A
1008-2794(2015)04-0018-05
2016-04-02
国家自然科学基金理论物理专项基金项目“钙钛矿铁电体-半导体硅异质结的理论研究”(11347023)
于海林,副教授,博士,研究方向:计算物理、低维材料物理与器件,E-mail:yuhailin_79@cslg.cn.
猜你喜欢
强度与环境(2022年4期)2022-09-26
计算机应用与软件(2022年2期)2022-02-19
长沙大学学报(2019年5期)2019-11-12
陶瓷学报(2019年6期)2019-10-27
纤维复合材料(2018年2期)2018-12-07
军事文摘·科学少年(2017年4期)2017-06-20
湖南文理学院学报(自然科学版)(2016年3期)2016-08-16
上海航天(2014年1期)2014-12-31
应用化工(2014年8期)2014-08-08
遵义师范学院学报(2010年3期)2010-09-01
