
口-面-指综合征一家系的基因诊断
2016-10-20 12:07王建才高莹刘晓雁胡瑾瞿宇晋
中国麻风皮肤病杂志 2016年9期
王建才高 莹刘晓雁胡 瑾瞿宇晋
口-面-指综合征一家系的基因诊断
王建才1高莹1刘晓雁1胡瑾1瞿宇晋2
口-面-指(趾)综合征(oral-facial-digital syndromes,OFDS)是一种主要表现为面部畸形、口腔异常及骨骼畸形的先天性综合征,临床中极为少见,早期诊断比较困难。我们对临床所见的一例可疑患儿进行了全外显子测序,在其OFD1基因上发现了1个错义突变(c.2524 G>A),最终明确患儿的诊断,同时也发现其母亲携带相同的突变位点。
口-面-指综合征; OFD1基因; 基因诊断
口-面-指(趾)综合征(oral-facial-digital syndrome,OFD)是一种主要表现为面部畸形、口腔异常及骨骼畸形的先天性综合征。口腔畸形包括分叶舌、颊黏膜纤维束、不对称腭裂、上唇正中假裂、牙齿畸形等。该病临床上比较罕见,发病率大约为 1/ 250000[1],目前国内仅报道10余例,现介绍我科门诊诊治的一例OFD患者。
1 临床资料
患儿,女,外蒙古人,5岁9个月。因足部肿物畸形近5年来我院就诊。患儿一般情况可,智力发育及语言表达能力低于常人。体格检查:患儿眼距增宽,额骨突出,鼻翼软骨发育不良,略呈扁平状(图1a),左口角向下倾斜,口腔内牙齿发育不良伴部分缺如,上腭裂(5岁时已手术修补),下颌系带增生,面部及四肢可见轻度异色改变(图1b、c、d),手部可见小指与大拇指有多指畸形已手术(图1e),足部有多处瘢痕样肿物(图1f)。血常规、尿常规、心电图、听力检测未见异常。下肢及足部CT:右足拇趾趾骨增粗,趾间关节背屈;双足第1/2趾软组织分隔不全;右足第1、2趾、左足第4、5趾局部软组织增厚、突出体表;左足第5趾关节外侧可见一游离骨块。腹部B超:肝胆胰脾及双肾未见异常。
母孕期无特殊接触史及服药史,否认近亲结婚,否认家族遗传性疾病史,孕3产2,其中人工流产1胎。
根据患儿自幼发病的特点,就诊时以足部肿物、特殊面容以及多部位先天畸形为主要临床表现,初步考虑某种先天性疾病,为明确诊断,我们抽取患儿及父母的外周血各4 mL,进行全外显子测序,发现患儿的OFD1基因在2524位碱基发生G→A(c.2524 G>A)杂合突变,导致OFD1基因所编码的蛋白第842位氨基酸发生甘氨酸到精氨酸的错义突变(p.G842R)(图2a、b、c),根据基因检测结果,结合患儿特殊的面部表现、口腔内发育畸形及手指(足趾)畸形的表现,最终明确诊断为口-面-指(趾)综合征。
2 讨论
OFD又名Papillon-Leage-Psaum综合征[1],目前该病依据临床表现分为13个类型,其中以I型(即oral-facial-digital syndrome type1,OFD1)及II型较常见。该病最早于1941年由Mohr报道,1954年由法国人Paillion-Leage和Psaum再次报道了类似病例,因为报道的患者均为女性,当时推测该病为X染色体连锁显性遗传[2],而在之后40多年,Feather等[3]通过对两个伴发多囊肾的OFD家系进行基因多态性分析,将致病基因定位于Xp22,直到2001年Ferrante等[4]才最终证实Cxorf5是OFD I的致病基因。目前认为该病是一种以黏膜组织异常及多系带为特征的先天畸形,同时伴有舌面发育异常及颅骨发育不全。这一综合征主要表现三大特征:(1)面部畸形表现为额骨、顶骨隆起,眶距过宽,眼眦易位,鼻翼软骨发育不全,鼻尖扁平,皮脂腺、毛发异常等。(2)口腔畸形表现为系带异常、颊系带增多、增生,舌系带过短,非对称性腭裂,分叉舌,舌部错构瘤,多生牙、错位牙,釉质发育异常,小下颌等;(3)其他方面[5-7]如分叉、并、弯、多指(趾)畸形,手、足部骨骼呈后短状,指(趾)骨质疏松,先天性心脏病,多囊肾,卵巢和肝囊肿以及学习困难,语言障碍等。该患者临床表现为眶距过宽、额骨突出及鼻翼软骨发育不良,口腔内上颚缺损、牙齿发育不良,手足多个并指及多指畸形,具备该综合征的三大典型特征。
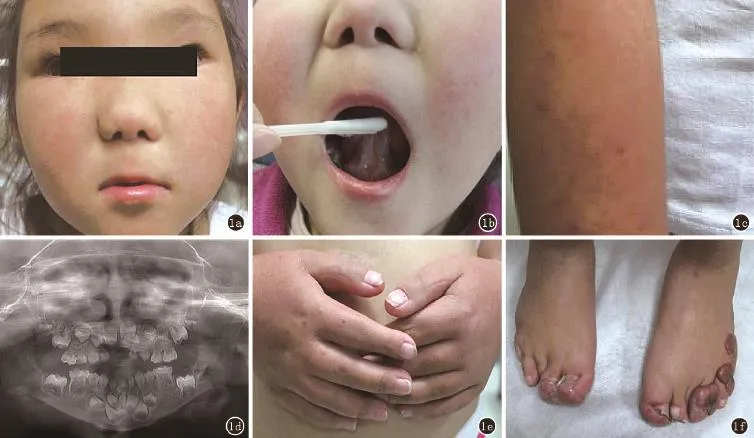
图1 1a患儿眼距增宽,额骨突出,鼻翼软骨发育不良,略呈扁平状;1b下颌系带增生;1b,c面部及上肢可见轻度异色改变;1d口腔内牙齿发育不良伴部分缺如;1e小指与大拇指有多指畸形(已手术);1f足部有多处瘢痕样肿物
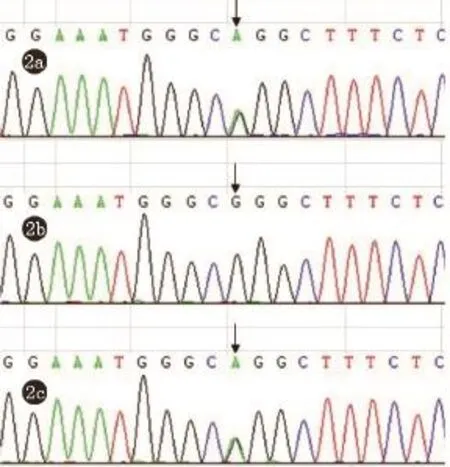
图2 患儿及双亲全外显子测序:患儿及其母亲OFD1基因在2524位碱基发生G→A(c.2524 G>A)杂合突变
OFD I型是X连锁的常染色体显性遗传性疾病,该突变对纯合男性个体具有致死性,故临床报道的存活患者基本都是女性,这点也符合其他X连锁显性遗传性疾病,比如色素失禁症。II型(Mohr综合征)为常染色体隐性遗传,主要特征性表现是口腔颌面部的舌裂及赘生物[8],无头发皮肤的异常,双侧拇指多指较常见,常伴传导性听力障碍,这些与本病例不符。结合以前的文献报道发现,并不是所有的I型患者都出现或者同时出现口腔、面部、指(趾)等部位的临床表现,文献报道约有50%为不完全表现型[9],除此之外有40%的患者可以出现神经系统异常[10],包括脑积水、胼胝体发育不全、后颅窝畸形和精神发育迟滞等。
由于该病比较罕见,不同个体间的临床表现差异较大,早期难以通过临床表现做出明确诊断,从而错失早期干预治疗,该患儿自幼已就诊多年均未能明确病因,来我科就诊后,根据患者病史及特殊体征考虑为某种先天性疾病,通过对患儿进行全外显子测序,最终发现了患儿的致病基因,做出明确诊断,给予对症处理,以期延缓预后。患儿的母亲存在相同的突变,但目前查体仅有轻度舌系带增厚,无多指并趾畸形,亦未发现腭裂存在,这些均反映该病临床表现的异质性。目前大多数学者对这一现象认可的解释是可能与女性23号染色体的随机失活有关,但也有研究表示这种随机的失活对这一现象有一定作用,但是存在其他更复杂的而没有被认知的机制[6]。以往报道认为,I型患者区分其他型的特征性病变是存在多囊肾[7,11],但在该患儿腹部B超中没有发现异常,可能与患儿年龄尚小有关,需要继续随访观察。
[1]陈书军,韩小宪,计宁,等.口-面-指综合征I型2例报道[J].中国美容医学,2009,9:1275-1277.
[2]Gurrieri F,Franco B,Toriello H,et al.Oral-facial-digital syndromes:review and diagnostic guidelines[J].Am J Med Genet A,2007,143A(24):3314-3323.
[3]Feather SA,Woolf AS,Donnai D,et al.The oral-facialdigital syndrome type 1(OFD1),a cause of polycystic kidney disease and associated malformations,maps to Xp22.2-Xp22.3[J].Hum Mol Genet,1997,6(7):1163-1167.
[4]Ferrante MI,Giorgio G,Feather SA,et al.Identification of the gene for oral-facial-digital type I syndrome[J].Am J Hum Genet,2001,68(3):569-576.
[5]Baker LA,Agim NG.Nevus comedonicus in oral-facial-digital syndrome type 1:a new finding or overlapping syndromes[J].Pediatr Dermatol,2014,31(2):e48-51.
[6]Bisschoff IJ,Zeschnigk C,Horn D,et al.Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome:a study of the extensive clinical variability[J].Hum Mutat,2013,34(1):237-247.
[7]叶敏,江银华,闫俊杰,等.口-面-指综合征I型伴先天性心脑发育不全1例报道[J].口腔医学,2014,3:215-217.
[8]陈京奕,陈振东,张学强,等.Mohr综合征一例报告[J].华西口腔医学杂志,2002,20(6):457.
[9]马思维,文抑西.口-面-指(趾)综合征I型两例报告[J].中国美容医学,2007,2:220-222.
[10]Nanda A,Sharaf A,Alsaleh QA.Multiple milia in a newborn with congenital malformations:oral-facial-digital syndrome type 1[J].Pediatr Dermatol,2010,27(6):669-670.
[11]Feather SA,Winyard PJ,Dodd S,et al.Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease:clinical,radiological and histopathological features of a new kindred[J].Nephrol Dial Transplant,1997,12(7): 1354-1361.(收稿:2016-03-07 修回:2016-04-18)
Genetic diagnosis in a family with oral-facial-digital syndrome
WANG Jiancai1,GAO Ying1,LIU Xiaoyan1,HU Jin1,QU Yujin2.
1.Department of Dermatology,Pediatric Hospital Affiliated to Capitalinstitute of Pediatrics,Beijing 100020,China;2.Department of Medical Genetics,Capital Institute of Pediatrics,Beijing 100020,China Correspongding author:WANG Jiancai,E-mail:wangjiancai7772@163.com
Oral-facial-digital syndrome is a congenital syndrome,which is mainly manifested in the facial deformity,abnormal oral cavity and skeletal deformity.It is very rare,and difficult to be diagnosed. Through the whole exon sequencing,onemissense mutation(c.2524 G>A)was identified,so we finally confirmed the diagnosis of the child who was suspected as OFD,and also found her mother carried the same mutation.
oral-facial-digital syndromes;OFD1 gene;genetic diagnosis
首都儿科研究所青年科研基金(编号:青年-14-09)
1首都儿科研究所附属儿童医院皮肤科,北京,100020
2首都儿科研究所遗传实验室,北京,100020
王建才,E-mail:wangjiancai7772@163.com
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
天津医科大学学报(2021年4期)2021-08-21
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
学校教育研究(2019年19期)2019-11-23
中国生殖健康(2018年4期)2018-11-06
小天使·二年级语数英综合(2017年10期)2017-10-31
中国当代医药(2015年30期)2015-03-01
中国医疗美容(2015年5期)2015-02-03
湖北农业科学(2014年11期)2014-09-10
