
Bethlem肌病一家系病例报告并文献复习
2016-08-09 07:25罗月贝李秋香梁静慧毕方方张宁杨欢
中国神经精神疾病杂志 2016年5期
罗月贝李秋香梁静慧毕方方张宁杨欢
Bethlem肌病一家系病例报告并文献复习
罗月贝*李秋香*梁静慧*毕方方*张宁*杨欢*
目的本文报道一常染色体显性遗传的Bethlem肌病家系共三代人的临床表现、病理学和遗传学特点。方法采集家系成员临床资料,分析肌肉病理改变及行基因测序,并从伴关节挛缩和关节过松的肌病两方面讨论其鉴别诊断思路。结果该家系主要临床表现为肘、腕、远端指间关节及跟腱挛缩,远端关节松弛,近端肌无力及皮肤症状,肌肉病理表现为非特异性肌营养不良改变。结论本研究总结Bethlem肌病病诊断要点,达到提高临床医师对Bethlem肌病这一罕见病认识的目的。
Bethlem肌病VI型胶原蛋白病COL6A1
VI型胶原蛋白病由分别编码VI型胶原蛋白α 链3个亚基的COL6A1、COL6A2、COL6A3基因突变所致,临床表型包括Ullrich型先天性肌营养不良,中间型VI型胶原蛋白病和Bethlem肌病,以及肌硬化症[1]。VI型胶原蛋白病各亚型在我国仅有个例报道,发病率尚无结论[2-4]。本文报告我院收治的一Bethlem病家系的临床、肌肉病理表现及遗传学特点,并复习相关文献,总结该病诊断要点,提高临床医师对Bethlem肌病这一罕见病的认识。
1临床资料
1.1患者1(II:4)患者1即先证者,因“肢体无力20余年”来我院肌病门诊。患者为28岁已婚女性,从事销售业。父母非近亲结婚,患者为足月顺产第三胎,1岁2个月会行走,2岁会跑步,但跑跳均不如正常同龄人。患者自记事起即有远端手指无法完全伸直,但近端手指活动度大,可过度弯曲。10岁左右开始出现蹲起困难,15岁发现大腿前群肌肉萎缩,20岁后逐渐出现左上肢无法完全伸直,双上肢上抬重物费力及走路时双足无法完全着地,否认夜间呼吸困难等呼吸肌受累表现。
体格检查:患者一般生命体征平稳,双手掌侧皮肤柔软细腻,腹部可见一纵行剖宫产后瘢痕过度形成(图1B),双下肢伸面毛周角化,脊柱轻度侧突,双侧远端指间关节、腕关节、左侧肘关节、双侧跟腱挛缩,双侧远端手指无法伸直,双手合十无法完全并拢(Bethlem征,图1A)。神经科查体:精神、智力发育正常,中枢和周围神经查体无阳性发现。双侧股四头肌、胫前肌及腓肠肌萎缩。肌力检查:双侧闭目、鼓腮力稍差,颈曲4+级,颈伸5级,双侧肩外展4+级,肘伸4-级,肘曲4级,左侧腕伸、曲4+级,右侧腕伸、曲4级,双侧髋外展、内收、伸4+级,髋曲4-级,膝伸、曲4级,足背曲4-级,足跖曲4+级,无眼外肌、球肌及呼吸肌无力。四肢肌张力正常。深浅感觉正常,四肢腱反射可引出,病理征阴性,颈软,脑膜刺激征阴性。
实验室检查:血、尿、大便常规、肝、肾功能、血清电解质、凝血功能、心肌酶谱(其中肌酸激酶99U/L)均正常范围。肌电图示三角肌、肱二头肌、拇短展肌、股内侧肌运动单位电位波幅低,时限窄,多相波增多,呈病理/混合募集相。神经传导检查正常。心电图示正常窦性心律。
1.2患者2(I:2)及患者3(III:2)先证者母亲(I:2)就诊时52岁,自诉自幼跑跳尚正常,自30岁起逐渐出现蹲起、上楼、上举重物费力,无关节活动障碍,现可独立行走,生活可自理。除生育先证者外另有四次生育史,第一胎未满月时因“褥疮”去世,第二胎为死胎,先证者一弟一妹及其后代无类似肌无力或关节活动障碍症状(图2)。查体示颈曲3级,颈伸4+级,双侧肩外展、肘屈肌力4-/5级,肘伸4级,腕伸、腕曲4+级,髋屈3+级,髋伸4+级,膝伸、曲4-级,足背屈、跖曲4级,上肢伸面、下肢可见毛周角化。
先证者育二女一子(图2),均为足月顺产,其中长女及儿子无类似肌无力症状,8岁的二女儿自出生后即四肢松软,跑跳均较同龄儿差,3岁后出现双手腕关节无法完全伸直,否认心悸、夜间呼吸困难或短暂活动后气促等症状。查体示智力正常,肩胛带肌、骨盆带肌肌力4+/5级,远端5/5级。双侧腕关节轻度挛缩。
1.3肌肉活检及基因检测结果先证者肱二头肌活检肌肉病理:伊红-苏木素染色示肌纤维大小中度不等,小纤维多呈圆形,散在坏死及再生肌纤维,结缔组织中度增生,部分肌纤维被脂肪细胞取代。NADH染色示肌纤维内肌原纤维间网格状结构紊乱,可见分叶及虫噬纤维,余线粒体酶染色未见异常。糖原及脂滴无明显增多。萎缩肌纤维以1型纤维为主。抗dystrophin-N、-R、-C、抗sarco⁃glycan-α、β、γ、δ、抗dysferlin抗体免疫组织化学染色示肌细胞内相关蛋白表达均正常。患者2、3拒绝行肌肉活检。
1.4基因检测对先证者行神经肌肉病相关二代测序示COL6A1基因c.1056+1G>A杂合突变,La⁃mande等报道该突变可致Bethlem肌病[5]。患者父母及子女拒绝行基因检测。
2讨论
VI型胶原纤维普遍存在于肌肉、软骨、皮肤、肌腱、血管等组织中。VI型胶原蛋白病的病理机制包括线粒体通透转运孔异常开放、自噬缺陷和凋亡激活等[6]。研究发现不同COL6基因突变对VI胶原蛋白的合成、组装、生理功能有不同的影响。既往文献报道本例COL6A1c.1056+1G>A杂合突变可致转录子的第14号外显子被跳跃(图3)[5]。致病机制为该突变破坏了定义剪切位点的保守序列,剪切子无法识别该位点,从而导致下游外显子被跳跃,其编码的VI型胶原蛋白a1链三重螺旋区域内的18个氨基酸缺失。突变蛋白无法被正确组装,进而阻碍细胞分泌VI型胶原蛋白至细胞外基质。第14号外显子跳跃是Bethlem肌病中的最常见的突变类型[7]。携带该突变的患者症状较重,且症状常在40~50岁期间加速进展。

图1 A示Bethlem征,B示腹部剖腹产后异常瘢痕形成

图2 家系图

图3 先证者肌肉病理A:伊红-苏木素染色示结缔组织增生,脂肪组织取代肌细胞;B:NADH染色是虫噬纤维;C:酸性磷酸酶染色示除坏死肌纤维内酶活性明显增高外,其余“正常”肌纤维内酶活性亦有点状增高。Bar:50 mm;放大倍数:200×
在各VI型胶原蛋白病亚型中,Ullrich型先天性肌营养不良表型最重。患者表现为生后或婴儿期出现的轴性和近端关节挛缩、肌无力,以及远端关节过松,症状进行性发展,疾病后期患者多丧失行走能力,因呼吸肌无力多需呼吸机辅助通气。表型较轻的Bethlem肌病的遗传方式以常染色体显性为主,亦有部分患者因携带纯合或复合杂合突变发病。患者婴儿期至成年后出现缓慢进展的近端肌无力和脊柱侧突等关节畸形,可出现选择性指长伸肌挛缩所致的“Bethlem征”。与Ullrich型先天性肌营养不良不同的是,Bethlem肌病患者呼吸肌基本不受累。中间型VI型胶原蛋白病症状严重程度介于Ullrich型先天性肌营养不良及Bethlem肌病之间。肌硬化症为VI型胶原纤维病的一个特殊亚型,患者关节挛缩症状明显重于肌无力,肌肉呈坚硬的木质感。除了骨骼肌肉系统,VI型胶原蛋白病患者的症状还包括毛周角化和瘢痕异常形成等皮肤受累表现。
Ullrich型先天性肌营养不良和Bethlem肌病患者肌酸激酶多正常或轻度升高。骨骼肌核磁共振表现特点为除缝匠肌、股薄肌和长收肌外大腿肌群的弥漫受累,股直肌中央部分因明显脂肪组织浸润在T2上呈中央高信号,相较股外侧肌则为周边脂肪浸润明显而中央部分肌肉组织相对保留[8]。患者肌肉病理常规染色表现为无特异性的肌纤维大小不等、轻度肌纤维坏死、再生、结缔组织轻度增生等肌营养不良表现。肌肉的VI型胶原蛋白免疫组化染色可以从表达正常到完全缺失。
Bushby等[9]提出的Bethlem肌病的诊断标准包括如下几条:①家族史,常染色体显性或隐性;②新生儿特点,肌张力低,远端关节松弛,髋脱位,关节挛缩,脊柱侧弯;③发病年龄,儿童至青年;④运动功能,无或轻度运动发育迟滞,可行走,近端或全身肌无力,缓慢进展,肩胛带肌及下肢肌萎缩;⑤结缔组织受累,早期即可出现远端关节松弛,关节挛缩主要累及跟腱、指长屈肌、肘关节及脊柱;⑥皮肤表现,瘢痕过度增生,瘢痕疙瘩形成,关节伸面毛囊角化过度;⑦其他,无中枢神经系统或心脏受累表现,40~50岁前呼吸功能正常;⑧实验室检查,血肌酸激酶正常或轻度升高,肌源性或肌营养不良性肌肉病理改变,肌肉胶原蛋白VI染色多正常,而成纤维细胞胶原蛋白VI染色多异常;⑨肌肉磁共振示受累肌肉周边信号增高而中央相对正常,股直肌可见“中心影”征;⑩基因检测示COL6A1、COL6A2、COL6A3突变。
本文报道祖孙三代受累的一Bethlem肌病家系。其中先证者经基因检测确诊为COL6A1突变相关的Bethlem肌病,尽管无法明确母亲及女儿基因型,但其临床表现与患者类似且符合Bethlem肌病表现,故祖孙二人极可能携带该致病突变。该家系中先证者母亲表型最轻,表现为成年后起病、进展缓慢的近端肌无力,关节和皮肤受累症状并不明显;先证者的症状几乎囊括了所有Bethlem肌病的典型临床表现,包括幼儿期起病的近端肌无力,肘、腕、远端指间关节、跟腱和脊柱关节挛缩,掌指关节活动度大,双手掌侧皮肤柔软,毛周角化和过度瘢痕形成;孙女的肌无力症状出生后即出现,伴近端关节挛缩。由于既往研究并未发现COL6相关基因在传代过程中的不稳定现象,本家系肌病症状在下一代中提前出现并非提示遗传早现,而是体现了Bethlem肌病的临床表现在家族内的异质性[7]。
VI型胶原蛋白病的肌肉病理为非特异性的轻度肌营养不良改变,但根据其关节挛缩和关节松弛等特征性临床表现可进行鉴别(见表1、2)。

图4 A示COL6A1c.1056+1G>A突变,B示COL6A1前体mRNA正常剪切模式,C示突变(箭头)致14号外显子跳跃
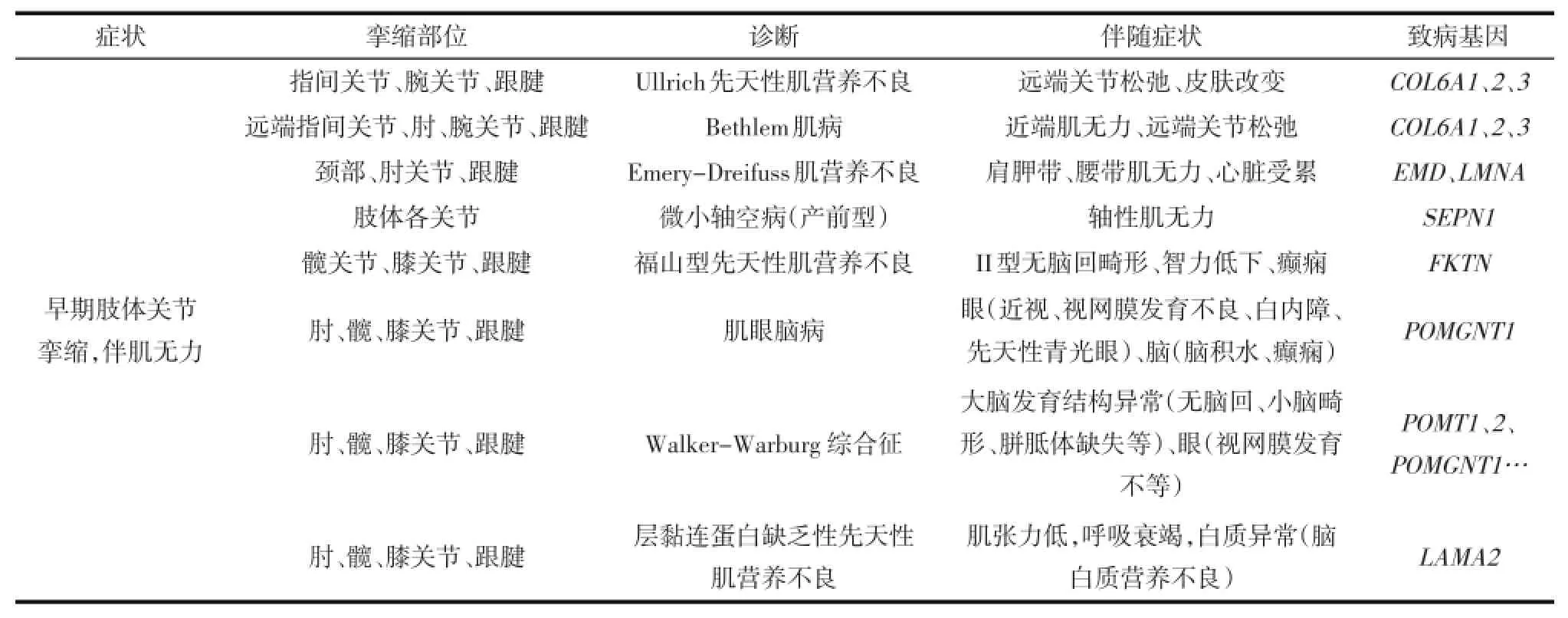
表1 伴关节挛缩的肌肉疾病鉴别诊断

表2 伴关节松弛的疾病相关鉴别诊断
本文报道COL6A1相关Bethlem肌病一家系,主要临床表现为肘、腕、远端指间关节及跟腱挛缩,远端关节松弛,近端肌无力及皮肤症状,临床表现在家族中有一定异质性。尽管症状与其他遗传性肌病或结缔组织病有一定相似处,但早期关节挛缩、远端关节松弛、近端型肌病和皮肤症状的临床表现组合高度提示VI型胶原蛋白病诊断。目前尚无针对VI型胶原蛋白病的特异性药物,对症治疗包括改善肌无力症状、适度功能锻炼延缓肌无力和关节挛缩进展、疾病后期呼吸机辅助通气等。
[1]BONNEMANN CG.The collagen VI-related myopathies:mus⁃cle meets its matrix[J].Nat Rev Neurol,2011,7(7):379-390.
[2]陆珺,朱雯华,卢家红,等.VI型胶原肌膜选择性缺失型Ull⁃rich型先天性肌营养不良临床及免疫病理特点[J].复旦学报(医学版),2009,36(4):454-456.
[3]蔡爽,朱雯华,陆珺,等.中国VI型胶原相关肌病的临床、病理和基因突变研究.中华医学会第十七次全国神经病学学术会议,2014:444-445.
[4]ZHANG YZ,ZHAO DH,YANG HP,et al.Novel collagen VI mutations identified in Chinese patients with Ullrich congenital muscular dystrophy[J].World J Pediatr,2014,10(2):126-132.
[5]LAMANDE SR,SHIELDS KA,KORNBERG AJ,et al.Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion [J].J Biol Chem,1999,274(31):21817-21822.
[6]BERNARDI P,BONALDO P.Mitochondrial dysfunction and de⁃fective autophagy in the pathogenesis of collagen VI muscular dys⁃trophies[J].ColdSpring HarbPerspect Biol,2013,5(5):a011387.
[7]DECONINCK N,RICHARD P,ALLAMAND V,et al.Bethlem myopathy:long-term follow-up identifies COL6 mutations pre⁃dicting severe clinical evolution[J].J Neurol Neurosurg Psychia⁃try,2015,86(12):1337-1346.
[8]MERCURI E,CINI C,PICHIECCHIO A,et al.Muscle magnet⁃ic resonance imaging in patients with congenital muscular dys⁃trophy and Ullrich phenotype[J].Neuromuscul Disord,2003,13 (7-8):554-558.
[9]BUSHBY KM,COLLINS J,HICKS D.Collagen type VI myopa⁃thies[J].Adv Exp Med Biol,2014,802:185-199.
(责任编辑:李立)
R746
A
10.3969/j.issn.1002-0152.2016.05.009*
中南大学湘雅医院神经内科
(E-mail:yangh69@yahoo.com)
2015-12-30)
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
中国医院院长(2021年21期)2022-01-21
今日农业(2021年5期)2021-11-27
山东医药(2021年24期)2021-09-01
中华养生保健(2021年18期)2021-02-13
医学信息(2020年12期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
诊断学(理论与实践)(2020年1期)2020-04-28
中国现代神经疾病杂志(2020年1期)2020-01-08
郑州大学学报(医学版)(2019年3期)2019-06-03
