
核磁共振波谱信号分析中的尺度缩放新算法
2016-08-04 08:29:22刘新卓邓伶莉王婉兰沈桂平许晶晶董继扬
厦门大学学报(自然科学版) 2016年4期
刘新卓,邓伶莉,2*,王婉兰,沈桂平,许晶晶,董继扬
(1.厦门大学 物理科学与技术学院,福建省等离子体与磁共振研究重点实验室,福建厦门361005;2.东华理工大学信息工程学院,江西南昌330013)
核磁共振波谱信号分析中的尺度缩放新算法
刘新卓1,邓伶莉1,2*,王婉兰1,沈桂平1,许晶晶1,董继扬1
(1.厦门大学 物理科学与技术学院,福建省等离子体与磁共振研究重点实验室,福建厦门361005;2.东华理工大学信息工程学院,江西南昌330013)
摘要:尺度缩放(scaling)是代谢组学数据处理中的一个重要环节,其主要目的是增强后续多元统计模型的分析能力.但目前常用的尺度缩放方法大多将尺度缩放当成一个独立的数据处理步骤,而未考虑多元统计模型的需要,使得后续的多元统计模型难以达到最优的分析能力.因此,提出一种模型自适应的数据尺度缩放算法,该算法将模型性能的代价函数写成变量缩放系数的一个函数,通过最大化代价函数来寻找最优的变量缩放系数.并且利用真实的核磁共振波谱数据对新方法进行评估,与单位方差法(unit variance,UV)、帕莱托法(Pareto)和变量稳定性法(variable stability,VAST)3种常用的尺度缩放方法进行比较.结果表明,新方法能够保留谱数据的结构信息,有效增强重要变量,抑制噪声和无关变量,提高模型的解释能力和预测能力.
关键词:尺度缩放;磁共振;波谱信号;多元统计分析
代谢组学方法是20世纪90年代末期发展起来的一门新兴科学,它借助于高通量、高灵敏度与高精确度的现代分析技术,分析细胞、组织和生物体液中内源性代谢物的整体组成,并通过代谢物复杂的、动态的变化,辨识和解析被研究对象的生理病理状态[1].在疾病早期诊断[2-3]、药物靶点分析[4]、环境毒理分析[5]等领域得到了广泛的关注.
核磁共振(nuclear magnetic resonance,NMR)波谱技术由于其非侵入性、无偏向性和可重现性等优点,已成为代谢组学研究中获取生物样品代谢物信息的重要手段之一[6-9].然而,在生物样品的NMR代谢波谱信号数据中,由于代谢物浓度的差别、氢核数目不同等原因,代谢物的信号强度往往相差很大.比如,正常人体尿液中肌酐(creatinine)和乳酸(lactate)的浓度相差近20倍,即使浓度相同的2种代谢物,由于其分子结构的差异、氢核数目的不同,对应共振峰的强度也会有很大差异.当利用主成分分析(principal component analysis,PCA)[10]、偏最小二乘分析(partial least square,PLS)[11]和正交偏最小二乘分析(orthogonal PLS,OPLS)[12]这类基于方差的多变量统计方法直接对这些数据分析时,弱信号的作用容易被强信号作用覆盖,而难于被发现[13].而实际上,强信号的变化可能只是由于尺度大造成的,不一定具有明显的生物学意义,因此为了消除数据尺度差异过大带来的不良影响,需要对数据尺度进行缩放处理[14].
在基于NMR的代谢组学中,常用的尺度缩放(scaling)方法主要有单位方差法(unit variance,UV)[15]、帕莱托法(Pareto)[16]和变量稳定性法(variable stability,VAST)[17].其中,UV方法处理后,各变量将具有相同的标准差.UV方法对噪声比较敏感,不利于特征信号的识别;Pareto方法是介于不做尺度缩放处理与UV方法之间的一种方法,在降低大信号的过重影响的同时一定程度上保持了原始数据的结构,相对于UV方法,得到的结果与原始数据更为相近;VAST方法的缩放“尺度”采用各组变量平均稳定性,由于噪声点的稳定性一般比较差,因此该方法可以有效降低噪声点的权重,改善UV方法的处理效果.尺度缩放的目的是为了改善多元统计分析效果,提高多元统计分析模型的可解释性.但是现有的这些尺度缩放方法只是从数据本身出发,并未考虑对后续多元统计分析的影响,因此,其多元统计分析(如PCA或PLS)的结果往往并不理想.
本研究提出一种模型自适应的尺度缩放方法(model adaptive scaling,MAS),即针对所采用的多元统计分析建模方法,给出适合该多元统计模型的最优尺度缩放方法.使用其对NMR检测的代谢指纹谱信号进行尺度缩放处理,能改善后续多元统计分析效果.
1理论与方法
设数据集中包含m个观测样本,每个样本有n个变量,则该数据集可用矩阵表示为X=(xij)m×n.尺度缩放方法可以用如下一般通式表示:

(1)
其中sj为矩阵X第j列(即第j个变量)的缩放系数.
UV方法采用各变量(列)的标准差作为缩放的“尺度”进行归一化,因此,数据经UV方法处理后各变量的标准差均为1.但由于信号采集过程中,噪声污染是不可避免的,而噪声的标准差通常较小,因此UV方法缩放后噪声相对于代谢物信号将被放大,不利于后续生物标志物的识别.Pareto方法将变量的标准差的开方作为“尺度”进行缩放,在一定程度上保留了原始数据的结构信息,同时也降低了大信号的过重影响,较UV方法的处理效果有所改善.VAST方法在UV方法的基础上,利用各变量在不同类别样本中的平均稳定性,进一步微调变量的缩放“尺度”,由于噪声信号的稳定性一般比代谢物信号的稳定性差,因此,VAST方法可以有效地降低噪声信号的权重,改善UV方法的处理效果.但VAST方法采用各组样本的平均稳定性,而某些代谢物虽然很稳定,但对于分组并无显著意义[13].例如:人体血液中肌酸(creatine)浓度相对稳定,但通常肌酸与大多疾病没有存在明显的关联.
尺度缩放是数据预处理的一个步骤,其目的是增强后续多元统计模型的分析能力.为了使后续的多元统计分析模型具有最优的分析能力,往往需要采用不同的数据预处理方法.然而,常用的UV、Pareto和VAST等方法均未考虑后续的多元统计模型的需要.针对这一问题,本研究提出了MAS方法.
设数据矩阵为X,样本响应矢量为Y,按如下步骤更新缩放系数s:
1) 初始化.分别对X和Y做中心化处理,并将设置s初始化值.
2) 模型建立.利用式(2)计算缩放后数据矩阵Xm;建立Xm的多元统计分析模型(如PCA或PLS),得到模型的负载矢量u.
Xm=X·diag(s).
(2)
3) 系数更新.更新缩放系数s,使得Xm在u上投影与响应Y线性相关性最大,即:
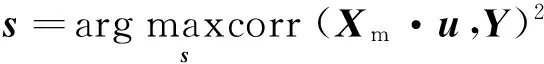
(3)
利用梯度下降法更新系数s:

(4)
其中相关系数r可以用Xmu和Y的协方差和标准差表示为
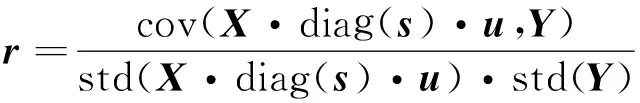
则有
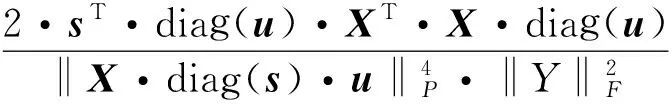
(5)
4) 停止条件.重复2)至3),直至s收敛,即Δs<ε.
上述算法中的负载u是由后续数据分析所选择的多元统计分析模型(如PCA或PLS)计算得到,选择不同的统计模型即可得到不同的u,称上述算法为MAS方法.缩放系数s反映变量的重要性,该算法是通过不断调整s来获得最终的收敛结果.考虑到s初始化可能对结果有一定的影响,本研究将s初始化为全1矢量.
2代谢指纹谱数据
为了验证MAS方法的有效性,采用该方法对实验获取的代谢指纹谱数据进行尺度缩放处理.实验数据来自一个关于素食人群代谢响应的研究,数据集由41个普通饮食男性志愿者(普食)和42个奶素食男性志愿者(素食)的尿液1H-NMR谱组成,素食志愿者均保持了5年的素食饮食习惯.关于样本收集的具体资料可参考文献[18].
尿液样本的1H-NMR谱数据均在Varian 500 MHz(Palo Alto,CA,USA)核磁共振谱仪上获得.数据预处理采用MetaboMiner V1.0软件[19]对谱图进行相位校正、基线校正和手动谱峰对齐等.取化学位移δ0.5~9.0区间的谱数据,并截除δ4.6~6.0(残余水峰和尿素峰)、δ0.6~0.8(DSS峰)、δ1.6~1.8(DSS峰)3个区域(DSS是2,2-二甲基2-硅戊烷-5-磺酸钠的简称),采用等间隔积分方法[20]将谱数据积分成1 348个数据点,然后利用组内聚合归一化(GAN)方法[21]对数据集进行归一化,用于降低尿液样品浓度差异对信号整体强度的影响,增强样本间的可比性.此外,发现普通组中有2个样本的葡萄糖信号远远大于其他样本,且在PCA得分图中这2个样本落入95%置信区间之外.推测这2个志愿者可能患有疾病,故将这2个样本剔除,最终得到一个81×1 348的数据矩阵X.
3实验与分析
3.1谱图结构分析
采用基于PCA模型的自适应尺度缩放方法(PCA-MAS)对数据矩阵X进行处理.为了比较分析,分别采用UV、Pareto和VAST方法对数据矩阵X进行尺度缩放,尺度缩放前后的堆积谱如图1所示,图中用不同颜色来区分不同样品的NMR谱.
图1(a)为原始数据的堆积谱,其中基线附近的小幅度变量主要由噪声数据点和一些浓度较低的代谢物共振峰组成,而幅度较大的变量则是高浓度代谢物的共振峰.在图1(a)中标记了2个肉眼能辨识的高强度共振峰A和B,用于观察不同尺度缩放方法对信号的影响.
图1(b)是UV方法缩放后的堆积谱,可见原始谱图中的基线的幅度被错误地放大了.代谢物的分子结构信息(即同一代谢物的不同共振峰之间的比例关系)被严重破坏,这可能使得后续的统计分析结果难于解释.此外,A和B峰完全淹没在周围噪声信号中,很难辨识.说明UV方法对噪声很敏感,不适用于低信噪比(SNR)的数据.
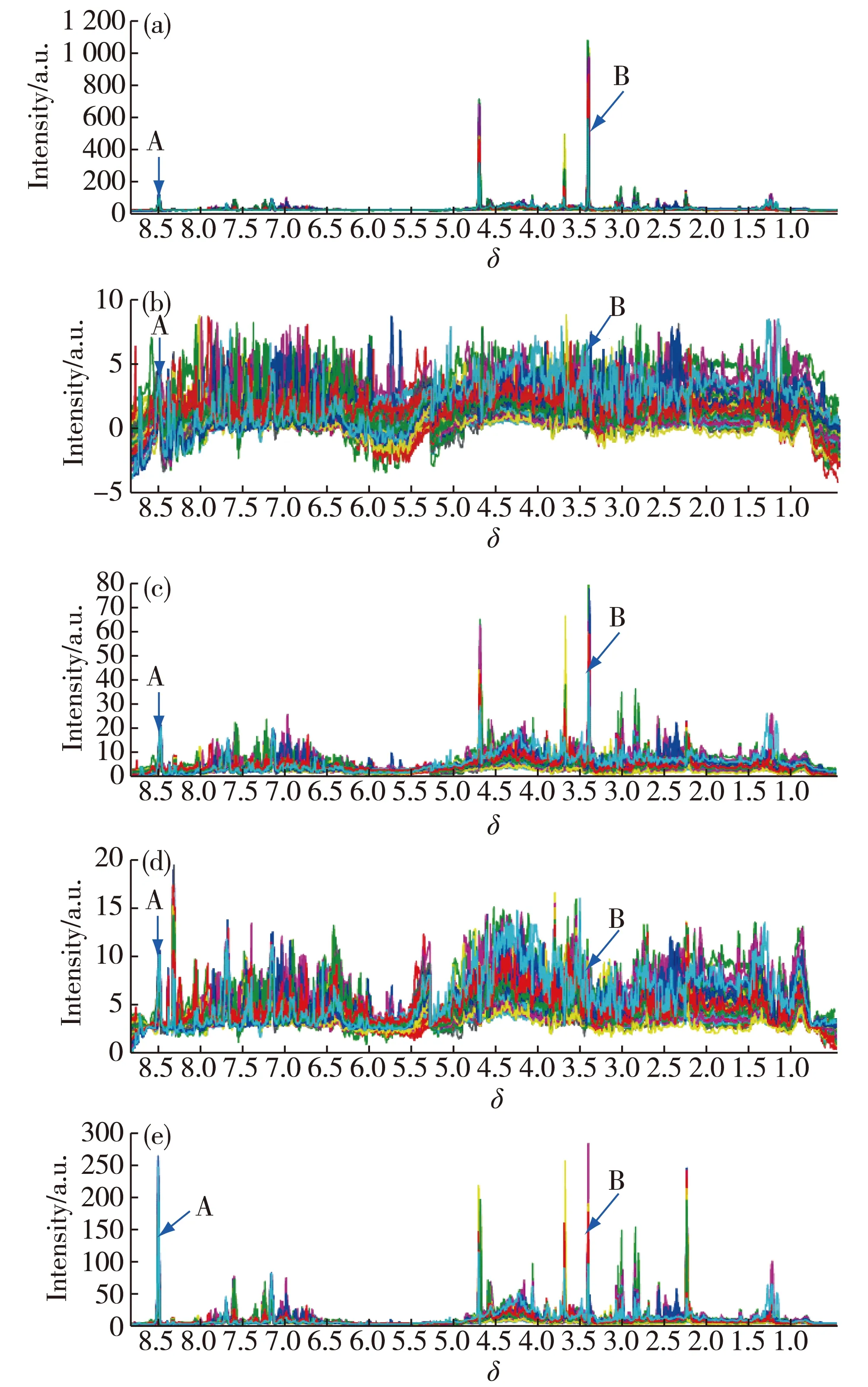
(a)原始数据;(b)UV;(c)Pareto;(d)VAST;(e)PCA-MAS.图1 尺度缩放后的堆积谱Fig.1Stacked spectrum processed by different scaling methods
图1(c)是Pareto方法缩放后的堆积谱,可以看出相对于UV方法的缩放结果,Pareto方法缩放对噪声变量有明显的抑制作用.2个标记谱峰与其邻近变量的对比度基本上得到了保持,但对标记强度较弱的信号峰A,其放大效果相对于被标记的较强的信号峰B并不明显.VAST方法是在UV方法的基础上对变量的权重进一步调整,由图1(d)中 2个被标记峰的缩放效果来看,虽然VAST与UV方法一样对谱图结构信息破坏严重,但还是能明显地识别出部分稳定性好的信号峰.
图1(e)是PCA-MAS缩放后的堆积谱,可以看出PCA-MAS对噪声信号的抑制效果是4种缩放方法中最好的,缩放后谱图中的谱峰信息得到了有效保留,肉眼很容易识别出.此外,对比2个被标记信号峰,较弱的信号峰A强度得到有效增强,同时较强的信号峰B得到了有效抑制.较强的信号峰和较弱的信号峰在缩放处理后,可比性较Pareto方法(图1(c))处理更好.
3.2统计建模分析
为了进一步对比尺度缩放效果,对各尺度缩放方法处理后的数据矩阵分别做PCA,对应的PCA得分图如图2所示.
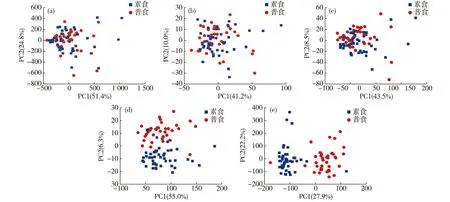
(a)原始数据;(b)UV;(c)Pareto;(d)VAST;(e)PCA-MAS. 横、纵坐标上的百分数为主成分贡献率.图2 数据尺度缩放后的PCA得分图Fig.2PCA score plots of datasets scaled by different methods
图2(a)~(c)中,原始数据、UV方法缩放和Pareto方法缩放后,素食和普食2组样本在PCA模型的前两个主成分上均无法区分开;VAST方法缩放后,2组样本在PCA第二主成分上能较好区分开,见图2(d);PCA-MAS方法缩放后,2组样本在PCA的第一主成分上可以明显的分开,而且较VAST方法具有较好的组内聚集性.这是由于素食和普食人群的差异变量(代谢物)的强度大多比较弱,对于原始数据这些弱的信号作用被强信号所掩盖,故PCA很难提取到;UV方法虽然能将所有变量缩放到同一个尺度(即具有相同的标准差),但是与此同时噪声信号也被放大,进而影响PCA对差异信息的提取;Pareto方法是权衡UV方法与不做缩放处理得到的一种折中方法,但弱的差异信号放大效果仍不够理想;VAST方法主要增强变异系数较小的变量,但变异系数小不代表这些变量对区分2组样本的贡献就大,因此VAST方法缩放后差异变量(即对区分2组样本的贡献较大的变量)所占的方差比例不一定最大,从而不能被PCA第一主成分所表征;而PCA-MAS方法对差异变量的增强作用比较好,对噪声或无关变量的抑制作用较强,因此,差异变量所占的方差比例较大,容易被PCA的第一主成分所表示.
3.3模型性能分析
结合蒙特卡罗交叉验证(Monte Carlo cross-validation,MCCV)[22]与受试者工作特性曲线下面积(area under a receiver operating characteristic curve,AUC)[23],分别采用内部验证和外部验证方法对不同尺度缩放方法处理后的数据对应的模型的性能进行定量评估.
内部验证:利用数据矩阵X计算缩放系数,尺度缩放处理后数据矩阵为Xm,从Xm中随机挑选80%样本作为训练集建模,剩下20%样本作为测试集;对训练集作PCA,提取第一个主成分建立PCA模型;利用AUC衡量测试集在模型中的可分性(即模型区分2组样本的能力).随机重复40次实验,取平均值及其标准差,记作Qint.
外部验证:对于数据矩阵X,每次随机挑选80%样本作为训练集建模,剩下20%样本作为测试集;计算训练样本的尺度缩放系数矢量s,对尺度缩放处理后的训练样本作PCA,提取第一个主成分建立PCA模型;利用缩放系数矢量s对测试样本进行尺度缩放处理,利用AUC衡量测试集在PCA模型中的可分性.随机重复40次实验,取平均值及其标准差,记作Qext.采用尺度缩放方法后各模型的预测能力如表1所示.可以看出,利用UV、Pareto和VAST方法处理后,模型预测能力基本相同,较原始数据(不做处理)的结果并没有明显提高;采用PCA-MAS方法处理后,模型无论是内部验证还是外部验证的预测能力(Qint=94%;Qext=81%),均远高于其他3种方法以及原始数据对应模型的预测性能.该结果进一步证明了PCA-MAS方法能够避免噪声以及无关代谢物对模型的干扰,突出特征代谢物信号对模型的响应.
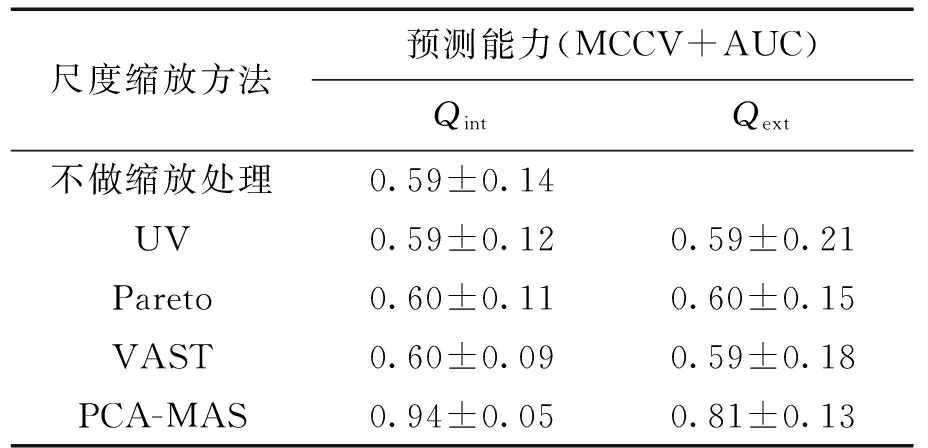
表1 不同尺度缩放方法比较结果Tab.1 Effect of different scaling methods
4结论
MAS能够针对所选取的多元统计分析模型,计算得到最优的尺度缩放系数来对数据进行尺度缩放处理,使得多元统计分析模型能准确提取特征信息.相对于以往基于数据的尺度缩放方法,该方法不仅能够有效保持谱数据的结构信息,抑制噪声以及强的非特征代谢物信号强度,同时能有针对性地提高重要代谢物信号的强度.另外,该方法对所采用的多元统计分析模型具有很好的适用性,使用灵活,处理结果有效.
参考文献:
[1]NICHOLSON J K,LINDON J C,HOLMES E.′Metabonomics′:understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data[J].Xenobiotica,1999,29(11):1181-1189.
[2]GOWDA G A N,ZHANG S C,GU H W,et al.Metabolomics-based methods for early disease diagnostics[J].Expert Review of Molecular Diagnostics,2008,8(5):617-633.
[3]RITCHIE S A,AHIAHONU P W,JAYASINGHE D,et al.Reduced levels of hydroxylated,polyunsaturated ultra long-chain fatty acids in the serum of colorectal cancer patients:implications for early screening and detection[J].BMC Medicine,2010,8(1):13.
[4]CLAYTON T A,LINDON J C,CLOAREC O,et al.Pharmaco-metabonomic phenotyping and personalized drug treatment[J].Nature,2006,440(7087):1073-1077.
[5]BUNDY J G,SPURGEON D J,SVENDSEN C,et al.Environmental metabonomics:applying combination biomarker analysis in earthworms at a metal contaminated site[J].Ecotoxicology,2004,13(8):797-806.
[6]GRIFFIN J L,WILLIAMS H J,SANG E,et al.Metabolic profiling of genetic disorders:A multitissue1H nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue[J].Analytical Biochemistry,2001,293(1):16-21.
[7]OWUSU-SARFO K,ASIAGO V M,DENG L,et al.NMR-based metabolite profiling of pancreatic cancer[J].Current Metabolomics,2014,2(3):204-212.
[8]JIMENEZ B,MIRNEZAMI R,KINROSS J,et al.1H HR-MAS NMR spectroscopy of tumor-induced local metabolic "field-effects" enables colorectal cancer staging and prognostication[J].Journal of Proteome Research,2013,12(2):959-968.
[9]危阳洋,王彩虹,李伟,等.甲亢患者血清和尿液的核磁共振代谢组学研究[J].高等学校化学学报,2010,31(2):279-284.
[10]WOLD S,ESBENSEN K,GELADI P.Principal component analysis[J].Chemometrics and Intelligent Laboratory Systems,1987,2(1/2/3):37-52.
[11]BARKER M,RAYENS W.Partial least squares for discrimination[J].Journal of Chemometrics,2003,17(3):166-173.
[12]BYLESJO M,RANTALAINEN M,CLOAREC O,et al.OPLS discriminant analysis:combining the strengths of PLS-DA and SIMCA classification[J].Journal of Chemometrics,2006,20(8/9/10):341-351.
[13]董继扬,李伟,邓伶莉,等.核磁共振代谢组学数据的尺度归一化新方法[J].高等学校化学学报,2011,32(2):268-274.
[14]温锦波,杨叔禹,肖娴,等.基于核磁共振的代谢组学数据预处理[J].厦门大学学报(自然科学版),2007,46(6):783-787.
[15]VAN DEN BERG R A,HOEFSLOOT H C J,WESTERHUIS J A,et al.Centering,scaling,and transformations:improving the biological information content of metabolomics data[J].Bmc Genomics,2006,7(4):1-15.
[16]ODUNSI K,WOLLMAN R,AMBROSONE C,et al.Detection of epithelial ovarian cancer using1H-NMR-based metabonomics[J].International Journal of Cancer,2005,113(5):782-788.
[17]KEUN H C,EBBELS T M D,ANTTI H,et al.Improved analysis of multivariate data by variable stability scaling:application to NMR-based metabolic profiling[J].Analytica Chimica Acta,2003,490(1/2):265-276.
[18]XU J J,YANG S Y,CAI S H,et al.Identification of biochemical changes in lactovegetarian urine using1H NMR spectroscopy and pattern recognition[J].Analytical and Bioanalytical Chemistry,2010,396(4):1451-1463.
[19]董继扬,周玲,邓伶莉.代谢组学数据挖掘软件(简称:MetaboMiner V1.0):2013SR060215[Z].2013-06-21.
[20]DIETERLE F,ROSS A,GÖTZ SCHLOTTERBECK A,et al.Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures.Application in1H NMR metabonomics[J].Ana-lytical Chemistry,2006,78(13):4281-4290.
[21]DONG J,CHENG K K,XU J,et al.Group aggregating normalization method for the preprocessing of NMR-based metabolomic data[J].Chemometrics and Intelligent Laboratory Systems,2011,108(2):123-132.
[22]XU Q S,LIANG Y Z.Monte Carlo cross validation[J].Chemometrics and Intelligent Laboratory Systems,2001,56(1):1-11.
[23]GREINER M,PFEIFFER D,SMITH R D.Principles and practical application of the receiver-operating cha-racteristic analysis for diagnostic tests[J].Preventive Veterinary Medicine,2000,45(1/2):23-41.
doi:10.6043/j.issn.0438-0479.201601006
收稿日期:2016-01-06录用日期:2016-03-08
基金项目:国家自然科学基金(81371639,31372546);福建省自然科学基金(2015Y0032);厦门大学校长基金(20720150018);东华理工大学科研基金(DHBK2015308)
*通信作者:denglingli1987@sina.com
中图分类号:O 658
文献标志码:A
文章编号:0438-0479(2016)04-0564-06
A New Variable-scaling Method for NMR Spectral Data Analyses
LIU Xinzhuo1,DENG Lingli1,2*,WANG Wanlan1,SHEN Guiping1,XU Jingjing1,DONG Jiyang1
(1.Fujian Provincial Key Laboratory of Plasma and Magnetic Resonance,College of Physical Science and Technology,Xiamen University,Xiamen 361005,China;2.School of Information Engineering,East China University of Technology,Nanchang 330013,China)
Abstract:Variable scaling,which aims to improve the performance of subsequently multivariate model,is a crucial procedure in metabolomic data analysis.However,most of the existing variable-scaling methods were carried out independently rather than taken into consideration the subsequently statistical model.Therefore,it is difficult for the optimal statistical model to be achieved.This paper proposes a model-adaptive-scaling method(MAS) for metabolic profiling analyses.The proposed method updates scaling coefficients of variables by upgrading the preselected statistical model.A real-world nuclear-magnetic-resonance-based(NMR-based) metabolic profiling set was used to evaluate the proposed method and to compare with other three commonly-used scaling methods,i.e.,unit variance(UV) scaling,Pareto scaling,and variable stability(VAST) scaling.Experimental results show that the proposed method outperforms other scaling methods in preserving the molecular information of spectra,enhancing important variables,and promoting the predicative ability of the preselected multivariate model.
Key words:scaling;magnetic resonance;spectral signal;multivariate statistical analysis
引文格式:刘新卓,邓伶莉,王婉兰,等.核磁共振波谱信号分析中的尺度缩放新算法[J].厦门大学学报(自然科学版),2016,55(4):564-569.
Citation:LIU X Z,DENG L L,WANG W L,et al.A new variable-scaling method for NMR spectral data analyses[J].Journal of Xiamen University(Natural Science),2016,55(4):564-569.(in Chinese)
猜你喜欢
中国临床医学影像杂志(2021年6期)2021-08-14 02:22:00
中国生殖健康(2020年6期)2020-02-01 06:29:06
中国生殖健康(2018年6期)2018-11-06 07:09:42
中国医药指南(2017年3期)2017-11-13 02:56:03
现代商贸工业(2017年5期)2017-03-29 19:20:01
中国经贸(2016年23期)2017-03-01 23:26:21
未来英才(2016年1期)2016-12-26 21:06:56
科技资讯(2016年19期)2016-11-15 10:06:37
商(2016年8期)2016-04-08 15:32:17
新校园·中旬刊(2014年11期)2015-01-12 13:29:19
