
新型降糖药利格列汀药理及光谱活性的密度泛函研究
2016-07-22 08:48:28卢巧君袁康杰方伯勇裴诗恩刘诗咏台州学院浙江台州318000
当代化工 2016年1期
关键词:利格列汀
陈 航,周 玥,卢巧君,袁康杰,方伯勇,裴诗恩,刘诗咏(台州学院,浙江 台州 318000)
新型降糖药利格列汀药理及光谱活性的密度泛函研究
陈 航,周 玥,卢巧君,袁康杰,方伯勇,裴诗恩,刘诗咏
(台州学院,浙江 台州 318000)
摘要:用密度泛函理论方法(DFT),模拟计算了利格列汀分子的分子-电子吸收光谱,并对其进行了指认。结果表明,利格列汀分子发挥药理活性的亲电和亲核反应中心很可能是氨基(NH2)和N17,而亲电中心则可能是羰基。
关键词:密度泛函理论;利格列汀;电子吸收图谱
糖尿病作为一种常见的慢性病,在市面上用于治疗该种疾病的药物多种多样,而利格列汀(Linagliptin)便是其中一种。德国勃林格殷格翰公司于2011开发的口服抗糖药利格列汀,它是一种二肽基肽酶-4(DPP-4)抑制剂,通过抑制体内DDP-4酶的活性来降低GLP-1的降解,而GLP-1在血糖控制和改善血糖水平上具有重要作用。利格列汀是首个主要经由胆道和胃肠道排泄的DPP-4抑制剂,给药剂量中仅有5% 经由肾脏排泄。无论患者肾功能处于何种水平,无需进行剂量调整。利格列汀代谢量极少,主要通过肠肝途径而不是肾脏途径进行排泄。 利格列汀可在所有患者人群中产生具有临床意义的、可靠的、持久的 HbA1c 改善作用,而且不受患者年龄、HbA1c 基线水平或获得确诊后的病程长短的影响。在临床医学上利格列汀能与多种药物联合使用,同时还具有和安慰剂相同的安全性与耐受性[ 1-4 ]。在服用利格列汀的过程中,应注意与饮食的相匹配,同时还要适量运动,这样才能更有效的改善2型糖尿病患者的血糖控制能力,起到更好的疗效。利格列汀为应对2型糖尿病患者肝肾功能减退,降低其后续的不良反应,提供了一个新的服药方式。
陈华妮等[5]采用分子对接原理对利格列汀分子的活性位点进行研究。随着量子理论和计算机技术水平的快速发展,人们已能计算机对量子力学进行简单的计算,而人们更希望能够利用计算机对微观体系中的量子力学方程进行求解 ,但是量子力学的基本方程—薛定谔方程的求解是极其复杂的。直到电子密度泛函理论 (DFT) 的建立,人们才克服了这一复杂性的难题[6,7]。本文采用密度泛函理论对利格列汀分子的红外,紫外可见,氢核核磁共振等分子-电子吸收光谱进行模拟和分析,取得了与实验相吻合的结果。从密度泛函理论入手,对格列卫分子在6-311+G(d,p)基组水平上,对格列卫分子进行初步的模拟研究,通过模拟得到一些相关数据比如NBO电荷等的基础上,对其易发生的反应部位进行了理论预测,具有降低科研成本方面的现实意义。
1 计算方法
本文对利格列汀分子(图1)采用密度泛函理论的BLYP方法,在6-31+G基组水平上对其分子骨架进行了结构优化与模拟。采用频率分析方法,对优化得到的稳定构型进行分析,得到的转-振动频率全部为正,表明我们的计算模拟方法是可性的。

图 1 利格列汀(C25H28N8O2)分子结构Fig.1 C25H28N8O2molecular structure
2 结果和讨论
2.1分子结构
用高斯程序在BLYP/6-31+G基准水平进行优化,对利格列汀分子与其晶体结构值比较示于表1。
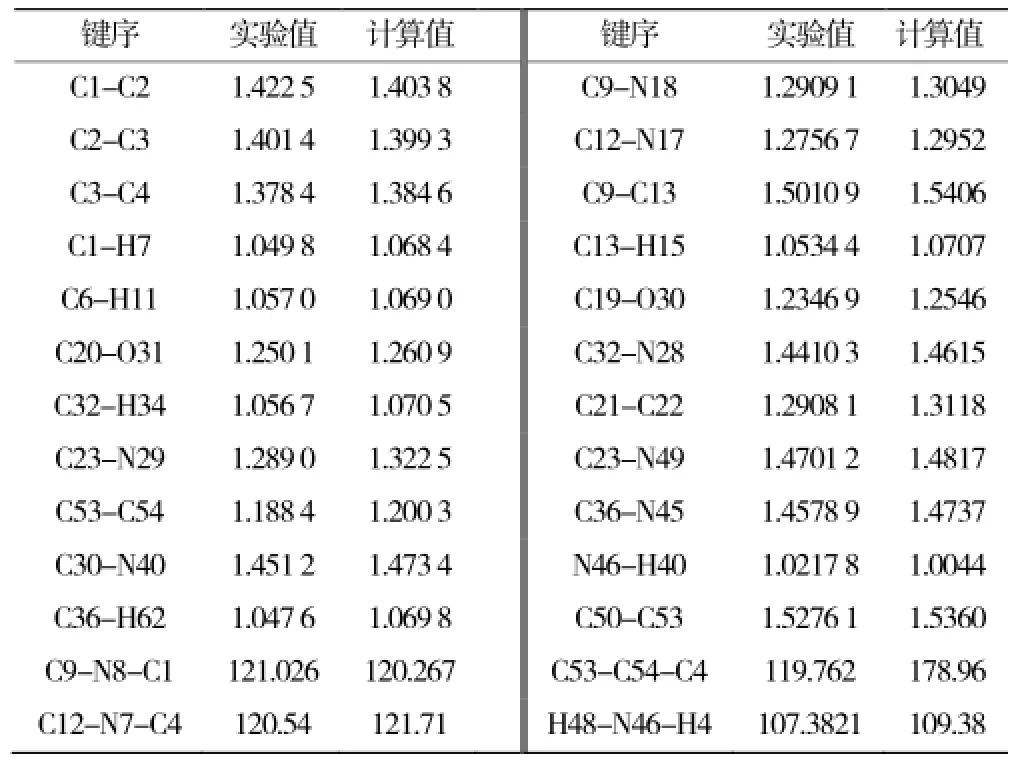
表1 分子的键长(Å)、键角值对照(°)Table 1 The optimized bond angels,bond lengths (nm,°)
正常的C-C 单键(1.54~1.62 Å),C-N单键(1.47~1.52 Å),C=C 双键(1.32~1.36 Å),C-O 单键(1.20~1.43 Å),C=O 双键(1.21~1.23 Å)。表1 中列出了优化后分子的部分键长数据表明∶ C1-C2、C2-C3稍短,C20-C31、C53-C54、C23-N29、O13-C14、C21-O22、C32=O28、C9=N18稍长,结合分子结构图1分析,符合离域结构稳定的要求。C36-C37-C38-C40-C42-N45六元杂环,键角均在110°左右,各原子之间的角度没有发生明显的扭曲变形。这些都有利于结构的稳定,位阻效应的减小及分子总能量的最低。
2.2紫外吸收光谱
采用TD DFT/BLYP/3-21G方法,模拟显示(见图2),利格列汀分子在533.21 nm (强吸收)和366.18 nm(弱吸收)处显示了紫外-可见吸收峰,533.21 nm处的吸收可归属为电子由最高占据轨道跃迁至最低空轨道,366.18 nm处的吸收峰可归属为电子从次高占据轨道跃迁至最低空轨道。这与其实验吸收谱图在520 nm处有较强的可见光吸收峰基本上是相吻合的。
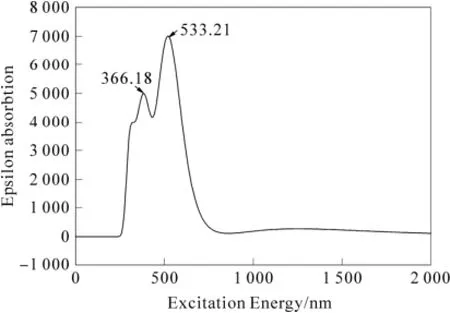
图2 利格列汀分子的紫外-可见吸收模拟光谱Fig.2 Simulation UV spectrum for Linagliptin
2.3红外吸收光谱(IR)
采用密度泛函理论方法 DFT/BLYP/3-21G模拟显示,利格列汀分子在2 062.36 cm-1(强,O-C伸缩振动),1 752.15 cm-1(中强,N-C,C-H伸缩振动),1 663.86 cm-1(中强,C-H面内摇摆振动),1 574 cm-1(中强,甲基和苯环C-H面内摇摆振动),282.017 cm-1(弱,甲基C-H面外面外摇摆振动吸收峰) 等处分别显示了红外吸收峰,这些峰与利格列汀分子实验红外吸收(IR)图谱在2 004.12,1 746.45,1 653.23,1 574.89,2 79,34 cm-1有较强的吸收峰是大致相吻合的(图3)。

图3 利格列汀分子的IR模拟图谱Fig.3 Simulation IR spectrum for Linagliptin
2.4拉曼吸收光谱
采用密度泛函理论 DFT/BLYP方法,在 3-21G基组水平上进行了分子构型的优化和频率分析,得到5个特征峰。对5个特征峰的分子振动形式进行了详细的归属指认。图4是模拟的利格列汀分子的拉曼散射谱特征峰,主要位于2 874.74 cm-1(碳碳三键上的碳对称伸缩振动),2081.2cm-1(六元环上两个羰基的对称伸缩振动),3 750.45 cm-1(苯环上的4个H的面内对称伸缩振动),3 899.07 cm-1(NH2上H的对称伸缩振动)和4 084.74cm-1((NH2上H的反对称伸缩振动)及范围内。优化计算的到的拉曼光谱与实验得到的吸收峰基本吻合。
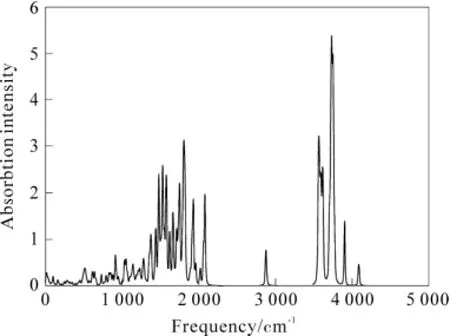
图 4 模拟利格列汀分子的拉曼图谱Fig.4 Simulation Raman spectra of Linagliptin
2.5核磁共振图谱(1HNMR)
氢原子具有一定的磁性,氢原子的原子核如果被电磁波照射,可以通过不同氢原子的这种吸收差异-化学位移进行理论模拟,利格列汀分子的氢核磁共振谱(1HNMR,见图5所示),其苯环上4个氢(弱,25~25.5×10-6),六元环上的 H(中或强,31.5~32.5×10-6);氨基上的H(弱,32.7×10-6);CH2上的H(弱,22.3 ×10-6) 。这些吸收峰也较好地与其实验值相吻合,也进一步验证了优化模拟计算的可行性。

图5 利格列汀的1HNMR模拟图谱Fig.5 Simulation1HNMR of Linagliptin
2.6 NBO电荷
表2的原子自然电荷(NBO)模拟数据值显示,氨基氮(N45)电荷最负(-0.414e),其次为与取代萘基上的碳的氮(N17,-0.255e);电荷最正的是羰基(O30)上的碳原子(C19,0.277e)。自然电荷计算表明,氨基(NH2)和N17很可能是其发挥药理和药理活性的亲电和亲核反应中心,而羰基很可能是亲电中。在模拟实验中,我们列出了格列卫分子相关原子电荷参数,其中原子自然原子轨道电荷值见表2所示和分子前线轨道图6。

表2 利格列汀分子的自然电荷分布值Table 2 Molecular natural charges of Linagliptin

图6 亲核(HOMO)和亲电(LUMO)轨道Fig.6 HOMO/LUMO for Linagliptin
3 结 论
(1)模拟出的利格列汀紫外-可见光吸收峰与实验值基本吻合验证了我们的研究方法的可行性;
(2)模拟的利格列汀红外光谱(特别是几处有强吸收的峰)所对应的键或基团在格列卫分子中都能找到,进一步论证了我们的研究方法可行性;
(3)利格列汀自然原子电荷值(NBO)及HOMO 和LUMO轨道模拟计算表明,利格列汀分子发挥药理活性的亲电和亲核反应中心很可能是氨基(NH2)和N17,而亲电中心则可能是羰基。这与陈力妮等人利用分子对接原理得到的利格列汀3个反应活性位点中的两个相吻合。
参考文献:
[1]钱荣立,项坤三,陈家伟,等.中国糖尿病防治指南[M].北京:北京医科大学出版社,2004:28-30.
[2]张儒雅,陆菊明.DPP4抑制剂在2型糖尿病治疗中的应用[J].中华内分泌代谢杂志,201l,27(1):43-47.
[3]范 鸣.抗2型糖尿病药Linagliptin[J].药学进展,2010,34(9):429-436.
[4]陆菊明.沙格列汀的临床研究进展[J].中国糖尿病杂志,2012,20(4):316-32.
[5]钱 力,陈华妮,杨文沛,等.linagliptin与靶标DPP4作用机理及分子设计研究[J].右江名族医学院,2013,41(5)∶13-15.
[6]骆天虹.GLP-1受体激动剂与二肽基酶4抑制剂治疗2型糖尿病的比较[J].中华内分泌代谢杂志,2012,28(9)∶44-48.
[7]黄占波,王丹,刘婷立,宋冬梅.抗2型糖尿病新药利拉利汀.实用药物与临床,2012,15(8)∶514-518.
Density Functional Study on Pharmacological and Spectrum Activity of Linagliptin as a New Hypoglycemic Drug
CHEN Hang,ZHOU Yue,LU Qiao-jun,YUAN Kang-jie,FANG Bo-yong,PEI Shi-en,LIU Shi-yong
(Taizhou University,Zhejiang Taizhou 318000,China)
Abstract:Molecular-electronic absorption spectra of Linagliptin molecules was simulated and calculated by density functional theory,and it was identified.The results show that,Linagliptin electrophilic molecules play a nucleophilic reaction center,and is likely to be pharmacologically active amino group (NH2) and N17,and the electrophilic center may be a carbonyl group.
Key words:DFT;Linagliptin;Electrocal absorption spectrum
中图分类号:O 644.32
文献标识码:A
文章编号:1671-0460(2016)01-0057-03
基金项目:国家级大学生创新训练计划项目,项目号:201510350015。
收稿日期:2015-09-30
作者简介:陈航(1994-),男,浙江义乌市人,现从事生物计算工作。 E-mail:347898183@qq.com。
通讯作者:刘诗咏(1978-),男,博士,研究方向:生物化学。
猜你喜欢
健康大视野(2020年21期)2020-12-30 14:03:34
糖尿病新世界(2019年10期)2019-08-27 00:00:00
糖尿病新世界(2019年10期)2019-08-26 01:33:09
糖尿病新世界(2019年9期)2019-07-29 00:40:44
中国实用医药(2019年9期)2019-07-01 13:46:39
糖尿病新世界(2019年23期)2019-02-10 04:01:13
糖尿病新世界(2018年15期)2018-12-10 10:40:28
糖尿病新世界(2018年13期)2018-12-10 09:15:10
当代医药论丛(2017年22期)2017-04-12 06:30:01
哈尔滨医药(2015年3期)2015-12-01 03:57:43
