
高通量测序分析1株H7N9流感病毒的全基因
2016-07-19 01:45:48王楷宬庄青叶张笑春陈继明
中国动物检疫 2016年4期
王楷宬,邱 源,庄青叶,张笑春,王 通,陈继明
(中国动物卫生与流行病学中心,山东青岛 266032)
高通量测序分析1株H7N9流感病毒的全基因
王楷宬,邱 源,庄青叶,张笑春,王通,陈继明
(中国动物卫生与流行病学中心,山东青岛 266032)
摘要:为探索利用高通量测序开展H7N9亚型流感的监测、全基因分析和溯源研究的方法,使用Ion Torrent PGM测序仪对1株H7N9流感病毒进行了全基因测序,分析了此毒株的基因组特性和进化特征。结果显示,该毒株与2013年引起我国华东地区疫情的H7N9流感毒株的亲缘关系较近;纵观在不同年代和地点分离的H7N9流感病毒的6个内部基因,2013年疫情之前和之后的H7N9流感病毒存在明显差异。
关键词:H7N9流感病毒;高通量测序;基因组;分子演化
资助项目:科技部科技基础性专项(SQ2012FY3260033);中国动物卫生与流行病学中心创新基金(2015IF-0004FF)
H7N9 流感病毒属于正粘病毒科、甲型流感病毒属。与其他甲型流感病毒一样,H7N9 流感病毒为有包膜、分节段、单负链RNA 病毒,基因组包括8 个片段。根据HA 和NA 抗原性的差异,甲型流感病毒可分为若干亚型,迄今已发现18种HA亚型(H1~H18)和11种NA亚型(N1~N11)[1-3]。任何一对HA 和NA 均可组合成一个亚型, 如H1N1、H5N1、H7N9等。
甲型流感病毒的宿主广泛,在人类中流行的有H1N1、H2N2 和H3N2 亚型,禽类常感染H5N1、H5N9、H3N8、H9N2、H5N2和H6N5等亚型[4]。禽类携带的流感病毒偶尔可感染人类,现已证实能直接感染人类的禽流感病毒有H5N1[5]、H9N2[6]、H7N2[7]、H7N3[8]、H7N7[9]、H7N9[10]、H5N2[11]、H10N7[12]和H10N8[13]等。
自2013 年2 月19 日开始,我国东南部的上海、安徽等地陆续发现由甲型H7N9 流感病毒引起的人感染病例。全基因系统进化分析表明,此次引起人类感染的H7N9 流感病毒与以前在禽类中发现的H7N9流感病毒完全不同,是一种从未出现过的新型流感病毒[10]。分析H7N9流感病毒全基因,对了解H7N9流感病毒的遗传演化关系,以及监测该病毒的发生发展趋势有重要意义。因此,本文采用高通量测序方法对1株在我国华东地区分离到的H7N9流感病毒进行了全基因测序与分析。
1 材料和方法
1.1材料和仪器
PathAmp FluA Reagent Kit、Ion Xpress Plus Fragment Library Kit、Ion PGM Template OT2 200 Kit、Ion PGM Sequencing 200 Kit V2、E-Gel SizeSelect 2% Agarose、Ion 318 Chip Kit V2、 Dynabeads MyOne Streptavidin C1 Beads、Ion Xpress Batcode Aaptors、Qubit核酸浓度测定仪及配套试剂,均购自Life technologies公司;超净工作台(美国Forma Scientifi c);移液器(Eppendorf);孵化器(德州诚信孵化设备有限公司);高速台式离心机(德国Heraeus Biofuge primoR);PCR扩增仪(Perkin Elmeter Gen Amp PCR System 9600)。
1.2毒株
H7N9流感病毒株A/chicken/China/028/2014,由中国动物卫生与流行病学中心禽病监测室保存,2014年1月分离自我国华东地区某活禽交易市场的鸡拭子样品,按农业行业标准(NY/T 772-2013)已鉴定为流感病毒H7N9亚型。
1.3流感病毒全基因扩增
按照说明书操作,使用QIAamp Viral RNA Mini Kit (Qiagen,德国)提取病毒RNA。应用PathAmp FluA Reagent Kit对病毒 RNA进行反转录,并扩增生成全基因组cDNA。使用Qubit核酸浓度测定仪及配套试剂测定DNA浓度。
1.5文库构建
将纯化后的H7N9流感病毒全基因cDNA稀释成35 μL含有200 ng DNA的溶液,使用Ion Xpress Plus Fragment Library Kit将病毒全基因组DNA打断为200 bp的片段,并加入含有Barcode的接头,构建为PGM测序仪可识别的DNA文库。
1.6测序
将DNA文库稀释为26 pM,应用Ion PGM Template OT2 200 Kit对DNA文库进行测序前的样品处理。将处理后的样品加样至Ion 318 芯片,置于PGM测序仪进行测序。测序序列经PGM自带的FluAnalysis(v4.0)软件进行序列拼接。
1.7分析与毒力或宿主嗜性相关的关键位点
使用Lasergene软件将A/chicken/China/028/ 2014的HA基因翻译为蛋白质序列,分析HA蛋白的裂解位点(第333~341位氨基酸)序列[14],以及第226位氨基酸是否由Gln替换为Leu[15],分析NS蛋白中结合CPSF的特异性残基是否为GLEWN[16]。
1.8分子进化分析
利用NCBI的Infl uenza Virus Resource数据库,搜索并下载其收录的,来自全球的H7N9亚型流感病毒全基因序列,在Linux 系统下,对同一时间、同一地点的毒株随机各抽选1株,并将各株病毒的同一基因节段分别与测序得到的序列一起使用MEGA6.0[17]进行对齐,同时加入有关2013年H7N9毒株进化分析文章中[10,18-19]提及的来源毒株A/duck/Zhejiang/12/2011(H7N3)、A/brambling/Beijing/16/2012(H9N2)和A/Baikal teal/Hongze/14/2005 (H11N9)的基因节段。 采用此软件计算得出构建进化树的最佳运算模型,绘制系统进化树。
2 结果与分析
2.1流感病毒全基因组扩增及文库构建
使用PathAmp FluA Reagent Kit扩增出目的条带,得到该病毒全基因的所有节段扩增产物,见图1。纯化后的cDNA浓度为49.1 ng/μL,经稀释后构建成含有Barcode接头的200 bp DNA文库。
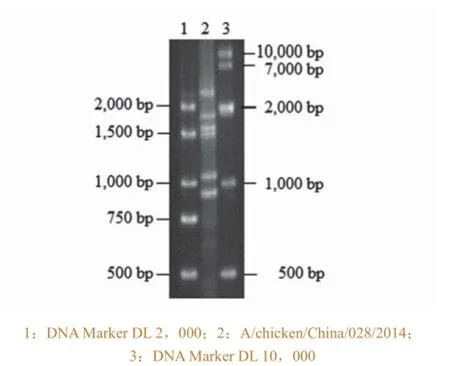
图1 PathAmp FluA Reagent Kit扩增A/chicken/ China/028/2014的全基因
2.2测序质量与测序深度
测序数据已上传至GenBank,登录号为SRR2970480。该病毒经基因组测序共产生193 448个reads,其中有187 117个reads正确匹配到甲型流感病毒的序列上,覆盖了流感病毒的所有8个节段。该毒株每个节段的平均测序深度见表1。包括各节段的5'和3'端序列在内的所有节段至少平均被测189次(PA基因)。NA基因的平均测序深度最大,为3 539。

表1 A/chicken/China/028/2014毒株各节段的测序深度
2.3与毒力或宿主嗜性相关的关键位点
该病毒HA蛋白的裂解位点(第333~341位氨基酸)序列为PEIPKGR↓GLF,为低致病性毒株。HA蛋白Q226为Leu,可紧密结合α-2和6 humanlike 受体,增加了通过空气传播的能力[15,20]。NS1蛋白能结合CPSF,其结合CPSF的特异性残基为GLEWN。该毒株特异性残基中的L突变为F,即GLEWN 突变为GFEWN,可能导致这些毒株致病力增强。
2.4病毒全基因分子进化分
该病毒8个节段的序列登录号为:KU318986-KU31进化分析发现,其与2013年引起我国华东地区疫情的H7N9流感病毒相似。HA基因和NA基因与来源于浙江的H7N3 流感病毒A/duck/Zhejiang/12/2011(H7N3)和韩国野鸟中发现的H7N9 流 感 病 毒A/wild bird/Korea/A14/2011(H7N9)亲缘关系近;PA、PB1、PB2和NP基因与来源于北京燕雀的H9N2 流感病毒株 A/ brambling/Beijing/16/2012 (H9N2)相近。该病毒的M基因、NS基因与2013年引起我国华东地区疫情的H7N9流感病毒存在一定的差异,其M基因与A/brambling/ Beijing/16/2012(H9N2)的同源性更高,NS基因与A/brambling/Beijing/16/2012 (H9N2)的亲缘关系稍远,均与2013年之后发现的,不同宿主的H7N9流感病毒同源。对不同时间、不同地区分离到的H7N9流感病毒的6个内部基因进行遗传进化分析发现,2013年H7N9疫情发生前与发生后的异,H7N9流感病毒的6个基因节支(图3)。仅1株2013年疫情uck/Zhejiang/LS02/2014(H7N9)其与2013年疫情之前的毒株亲缘关系稍远,与2014年疫情之后的毒株亲缘关系稍近。

图2 HA、NA基因分子进化分析
3 讨论
2013年春,在我国华东地区,H7N9流感病毒引起多人感染和死亡。Gao等[10]认为引起此次疫情的病原是一种三源重配的新型H7N9流感病毒。该病毒所有基因片段均来源于禽流感病毒,不含任何人流感病毒的基因片段,并且该病毒的8个基因节段来源于3个不同的禽流感病毒株。其中HA 基因与2011 年在我国浙江分离到的H7N3禽流感病毒株A/duck/ Zhejiang/12/2011(H7N3)同源性最高,NA 基因与2011年在韩国野鸟中发现的H7N9 流感病毒株A/wild bird/ Korea/A14/2011(H7N9)高度同源,其余6个内部基因与2012年在北京燕雀中分离到的H9N2禽流感病毒株A/brambling/Beijing/16/2012 (H9N2)同源性最高。虽然各基因片段分别与近年在东亚地区流行的禽流感病毒相近,但这种基因片段的组成此前从未在禽、人或其他动物中发现过。Xiong等[19]认为NA基因并非来源于韩国的野鸟,而是来自于江苏洪泽湖地区花脸鸭的H11N9 病毒A/Baikal teal/Hongze/14/2005(H11N9)。由于H7N9流感病毒为多源重配的新型流感病毒,且不同研究对其重组来源的分析结果不同,毒株的多样性也不十分清晰,因而对H7N9亚型流感病毒监测、全基因分析和溯源研究有重要意义。
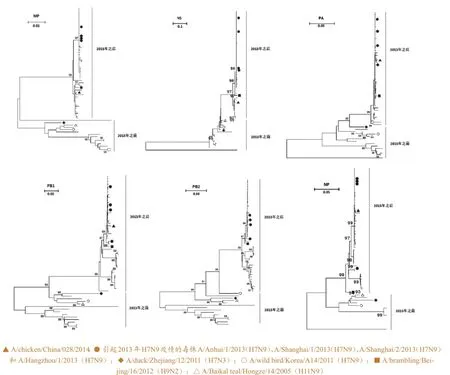
图3 病毒内部基因分子进化分析
本文采用Ion Torrent PGM高通量测序仪完成了1株H7N9流感毒株的全基因测序。结果显示,本文所采用的建库与测序方法能够测定流感病毒的全基因,并且测序深度较大,能满足序列分析的需要。本研究在一次测序中完成了该病毒全部8个基因的测序,能够满足对H7N9亚型流感监测、全基因分析和溯源研究的要求。全基因序列遗传进化分析发现,该病毒与2013年引起我国华东地区疫情的H7N9流感病毒的亲缘关系较近。针对Gao[10]和Xiong[19]对引起2013年疫情毒株的NA基因来源的争议,本文同时加入了A/wild bird/Korea/ A14/2011(H7N9)和A/Baikal teal/Hongze/14/2005(H11N9)两株病毒进行分析,结果发现本文测序的毒株和引起2013年疫情毒株的NA基因均与A/ wild bird/Korea/A14/2011(H7N9)相近,与Gao[10]的结论一致。从时间来看,纵观不同年代和地点分离到的H7N9流感病毒,以发生疫情的2013年为节点,H7N9流感病毒的6个内部基因分为两大分支。2013年之后的毒株不论宿主和地点绝大多数处于同一个大分支,各基因节段的亲缘关系较近,而且GenBank中2013年之后的毒株均分离自中国;而2013年之前的毒株都处于另一大分支。可见,H7N9毒株在2013年发生了对致病性有重要意义的重组和变异。
参考文献:
[1] Freidl G S,Binger T,Muller M A, et al. Serological evidence of infl uenza a viruses in frugivorous bats from Africa [J]. PLoS One,2015,10(5):e0127035.
[2] Tong S,Li Y,Rivailler P,et al. A distinct lineage of infl uenza A virus from bats [J]. Proc Natl Acad Sci U S A,2012,109(11):4269-4274.
[3] Tong S,Zhu X,Li Y,et al. New world bats harbor diverse infl uenza A viruses [J]. PLoS Pathog, 2013,9(10):e1003657.
[4] Avian infl uenza A(H5N1)--situation (poultry) in Asia as at 2 March 2004: need for a long-term response,comparison with previous outbreaks [J]. Wkly Epidemiol Rec,2004,79(10):96-99.
[5] Le M Q,Horby P,Fox A,et al. Subclinical avian infl uenza A(H5N1) virus infection in human, Vietnam [J]. Emerg Infect Dis,2013,19(10):1674-1677.
[6] Butt K M,Smith G J,Chen H,et al. Human infection with an avian H9N2 infl uenza A virus in Hong Kong in 2003 [J]. J Clin Microbiol,2005,43(11):5760-5767.
[7] Ostrowsky B,Huang A,Terry W,et al. Low pathogenic avian influenza A (H7N2)virus infection in immunocompromised adult,New York,USA,2003 [J]. Emerg Infect Dis,2012,18(7):1128-1131.
[8] Skowronski D M,Tweed S A,Petric M,et al. Human illness and isolation of low-pathogenicity avian infl uenza virus of the H7N3 subtype in British Columbia,Canada [J]. J Infect Dis,2006,193(6):899-900;author reply 900-891.
[9] Puzelli S,Rossini G,Facchini M,et al. Human infection with highly pathogenic A(H7N7) avian influenza virus,Italy,2013 [J]. Emerg Infect Dis,2014,20(10):1745-1749.
[10] Gao R,Cao B,Hu Y,et al. Human infection with a novel avian-origin infl uenza A(H7N9)virus [J]. N Engl J Med,2013,368(20):1888-1897.
[11] Ogata T,Yamazaki Y Okabe N,et al. Human H5N2 avian infl uenza infection in Japan and the factors associated with high H5N2-neutralizing antibody titer [J]. J Epidemiol,2008,18(4):160-166.
[12] Arzey G G,Kirkland P D,Arzey K E,et al. Infl uenza virus A (H10N7) in chickens and poultry abattoir workers,Australia [J]. Emerg Infect Dis,2012,18(5):814-816.
[13] Zhang T,Bi Y,Tian H,et al. Human infection with influenza virus A(H10N8)from live poultry markets,China,2014 [J]. Emerg Infect Dis,2014,20(12):2076-2079.
[14] Chen R A,Lai H Z,Li L,et al. Genetic variation and phylogenetic analysis of hemagglutinin genes of H9 avian influenza viruses isolated in China during 2010-2012 [J]. Vet Microbiol,2013,165(3-4):312-318.
[15] Imai M,Watanabe T,Hatta M,et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets [J]. Nature,2012,486(7403):420-428.
[16] 李建丽,陈恩林,李和平,等. H9N2亚型禽流感病毒NS1基因的进化分析 [J]. 病毒学报, 2008,24(3):220-226.
[17] Tamura K,Stecher G,Peterson D,et al. MEGA6:Molecular Evolutionary Genetics Analysis version 6.0 [J]. Mol Biol Evol,2013,30(12):2725-2729.
[18] Liu D,Shi W,Shi Y,et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection:phylogenetic,structural,and coalescent analyses [J]. Lancet,2013,381(9881): 1926-1932.
[19] Xiong C,Zhang Z,Jiang Q,et al. Evolutionary characteristics of A/Hangzhou/1/2013 and source of avian influenza virus H7N9 subtype in China [J]. Clin Infect Dis,2013,57(4):622-624.
[20] Herfst S,Schrauwen E J,Linster M,et al. Airborne transmission of influenza A/H5N1 virus between ferrets [J]. Science,2012,336(6088):1534-1541.
(责任编辑:朱迪国)
中图分类号:S855.3
文献标识码:B
文章编号:1005-944X(2016)04-0072-05
DOI:10.3969/j.issn.1005-944X.2016.04.024
通讯作者:陈继明
Genome Analysis of a H7N9 Infl uenza Virus by High-throughput Sequencing
Wang Kaicheng,Qiu Yuan,Zhuang Qingye,Zhang Xiaochun,Wang Tong,Chen Jiming
(China Animal Health and Epidemiology Center,Qingdao,Shandong 266032)
Abstract:To discuss the serveillance,genome analysis and source tracing of H7N9 infl uenza virus genome by highthroughput sequencing,the genome of a H7N9 infl uenza virus was sequenced by Ion Torrent PGM. The characteristics of genome and evolution were analyzed. The results showed that the virus was similar to those viruses causing the H7N9 infl uenza outbreaks of eastern China in 2013. Comprehensive analysis with the 6 internal genes of H7N9 infl uenza virus isolated from different areas and time showed that the H7N9 infl uenza viruses isolated after the outbreaks in 2013 were very different from those viruses isolated before.
Key words:H7N9 infl uenza virus;high-throughput sequencing;genome;molecular evolution
猜你喜欢
今日农业(2021年11期)2021-08-13 08:53:24
中西医结合肝病杂志(2020年2期)2020-10-27 02:18:50
中成药(2018年7期)2018-08-04 06:04:22
中国医药导报(2017年9期)2017-05-11 18:44:13
中国中药杂志(2016年24期)2017-04-18 17:42:52
中国中药杂志(2017年4期)2017-03-28 17:06:08
中国中药杂志(2017年3期)2017-03-20 21:11:11
科技创新导报(2016年28期)2017-03-14 11:32:29
江苏农业科学(2014年10期)2014-11-22 19:57:52
遗传(2014年2期)2014-02-28 20:58:18
