
多穗柯转录组分析及黄酮类化合物合成相关基因的挖掘
2017-03-28 17:06宋菊黄剑李志栋龙月红邢朝斌
中国中药杂志 2017年4期
宋菊+黄剑+李志栋+龙月红+邢朝斌
[摘要] 多穗柯具有甜味和保健功效,其中黄酮类化物是主要活性物质。为获取多穗柯转录组数据库以及黄酮类化合物生物合成相关基因,该研究用RNA-Seq中的Illumina HiSeq 4000对多穗柯嫩叶进行转录组测序,共获得6 Gb数据,拼接后得到41 043条Unigene,与7个基因数据库进行比对,可归类于51个GO分类中,涉及到237个KEGG标准代谢通路。找到黄酮合成相关基因28条。通过MicroSatallite(MISA)软件分析筛选到18 161个SSR,其中单碱基重复最丰富,有7 346个。此研究得到大量转录本信息,为多穗柯的分子生物学研究提供了宝贵的转录组数据库资源。
[关键词] 多穗柯; 转录组; 高通量测序; 黄酮类化合物
[Abstract] The sweet taste and health effect of Lithocarpus polystachyus are mainly related flavonoid. To obtain Lithocarpus transcriptome database and flavonoid biosynthesis-related genes, the RNA-Seq techology (Illumina HiSeq 4000) was used to sequence its transcriptome. Six Gb database was assembled after assembly steps, and 41 043 of L. polystachyus unigenes were obtained. With blasting them with 7 data banks, all unigenes were involved in 51 GO-terms and 237 metabolic pathways. And furthermore 28 genes of the flavonoid biosynthesis-related were found. After using the MicroSatallite, 18 161 SSR were obtained, the single-nucleotide-repeated was the richest at 7 346. These data represent abundant messages about transcripts and provide valuable genome data sources in molecular biology of L. polystachyus.
[Key words] Lithocarpus polystachyus; transcriptome; high-throughput sequencing; flavonoids
多穗柯Lithocarpus polystachyrus Rehd,别名甜茶,是壳斗科石柯属常绿乔木[1]。因其具有强烈的甜味和保健功能而被广泛关注,其中二氢查耳酮类化合物根皮苷和三叶苷是其主要活性物质,具有降血糖、保肝、抗氧化、抗肿瘤等生物活性[2]。三叶苷的含量在多穗柯二氢查二酮苷类中最高,占95%,且根皮苷和三叶苷互为异构体。目前,仅有根皮苷受到较高关注,它的合成途径[3]、作用[4]等都已有相关报道,但是关于多穗柯中根皮苷和三叶苷的合成、代谢以及它们相互转化的途径尚不够明确。因此,利用转录组测序技术,对多穗柯转录组进行分析,找出黄酮类化合物合成相关基因,为后续对根皮苷和三叶苷的研究提供资源。
转录组是指特定生物体在某种状态下所有基因转录产物的总和,转录组研究属于功能基因组学研究的范畴,是连接基因组与蛋白质组的纽带[5]。它能反映生物个体在特定器官、组织或某一特定发育、生理阶段细胞中所有基因表达水平的数据。可用来比较不同组织或生理状况下基因表达水平差异,发现与特定生理功能相关的基因,推测未知基因[6]。因此,采用转录组测序技术,对多穗柯转录组进行测序分析,建立起转录组数据库,为多穗柯中黄酮类化合物二氢查耳酮类的根皮苷和三叶苷生物合成研究打下基础。
1 材料与方法
1.1 cDNA制备
以多穗柯嫩叶为本研究原材料提取植物总RNA,再逆转录为cDNA,用于构建转录组数据库。植物总RNA 提取试剂盒和逆转录试剂盒购自天根生化科技(北京)有限公司。
1.2 转录组数据的组装与分析
1.2.1 组装 采用Illumina HiSeq 4000测序技术平台的PE150技术进行测序,将得到的Raw Data进行过滤处理,获得高质量的Clean Reads,使用trinity软件进行组装,得到Unigene。
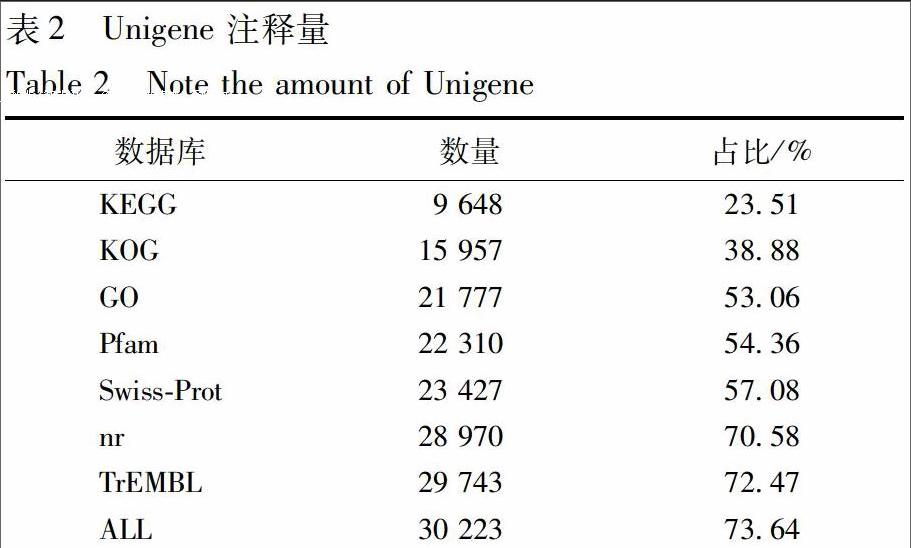
1.2.2 功能预测 利用BlastX将All-Unigene与nr[7],Swiss-Prot[8],GO[9],KOG[10],KEGG[11]等6个数据库进行比对。又使用KOBAS2.0[12]去获取序列在KEGG中比对KEGG Orthology结果,预测出序列的氨基酸序列之后,使用HMMER[13]软件与Pfam[14]数据库比对,获得注释信息。
1.2.3 黄酮类化合物合成相关基因的挖掘 根据康亚兰[15]和郭欣慰[16]提出的黄酮类化合物合成途径中的结构基因与调节基因,以及KEGG注释的结果和数据库中已知的基因信息,利用本地Blast進行检索比对,确定本转录组数据中与黄酮合成相关的基因。
1.2.4 SSR分析 微卫星序列(microsatellite DNA)又称为简单序列重复(simple sequence repeats,SSR) 或简单序列(simple sequences),是指以1~6个核苷酸为基本重复单位的串联重复序列,其长度大多在 100 bp 以内。它们广泛存在于各类真核生物基因组中,原核生物基因组中也含有少量的微卫星序列[17]。SSR作为分子标记的一种,被广发用于杂交育种、种群遗传多样性、遗传连锁图谱的构建等研究领域。目前关于多穗柯的分子标记十分有限,本研究利用MicroSatallite(MISA)软件找出全部的SSR,为多穗柯的遗传标记研究提供非常重要物质资源和依据。
2 结果与分析
2.1 组装
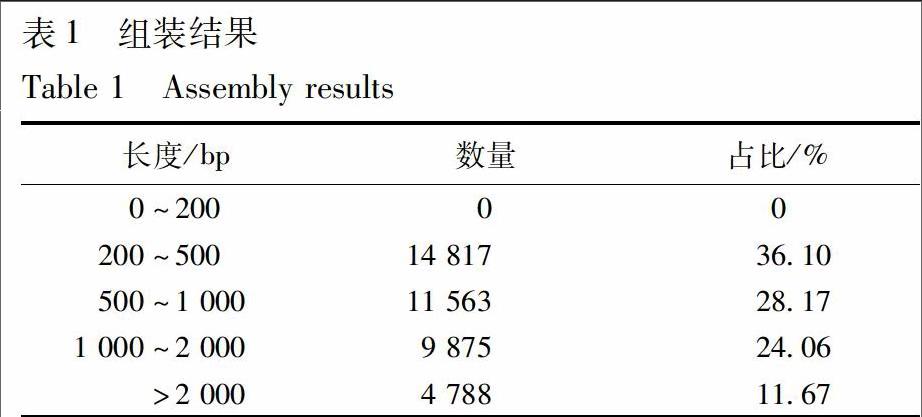
总共得到6 Gb的Clean Date,组装获得Unigene 41 043条,N50长度为1 472 bp,长度大于N50的Unigene 有8 977条,组装完整性较高,具体组装结果见表1。
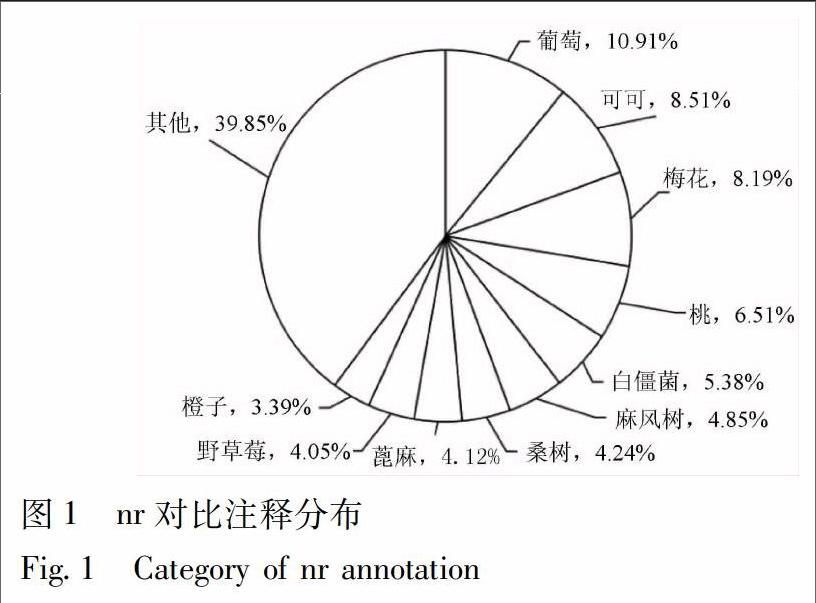
通过BlastX与nr数据库进行比对,有28 970 条Unigene获得注释,从匹配的物种来源分析,有10.91%的Unigene注释到葡萄中,8.51%注释到可可中,其余分别为梅花8.19%、桃6.51%、白僵菌5.38%、麻风树4.85%、桑树4.24%、蓖麻4.12%、野草莓4.05%,橙子3.39%,其余39.85%注释到其他物种中,见图1。
随后将所有的Unigene比对到KOG数据库中,结果显示有15 957条序列获得17 067个注释信息,划分为25个功能分类。从基因功能分布特征中可以发现一般功能预测基因分布最多,多达3 751条,涉及翻译后修饰、蛋白翻转、分子伴侣功能的基因次之,有1 736条,而涉及核结构、胞外结构和细胞运动的基因很少,仅有59条、56条和5条。此物种的KOG功能注释分布结构与其他物种不尽相同,见图2。
A.RNA加工和修饰;B.染色体结构和动力学;C.能源产生和转化;D.细胞周期调控,细胞分裂,染色体分离;E.氨基酸转运和代谢;F.核算转运和代谢;G.碳水化合物转运和代谢;H.辅酶转运和代谢;I.脂类转运和代谢;J.翻译,核糖体结构和生源;K.转录;L.复制,重组,修饰;M.细胞壁,细胞膜,被膜生源;N.细胞活性;O.翻译后修饰,蛋白反转,伴侣;P.无机离子;Q.次生代谢物的生物合成,转运和代谢;R.一般功能预测;S.未知功能;T.信号传递机制;U.细胞内运输,分泌和囊泡转运;V.防御机制;W.细胞外结果;Y.核结构;Z细胞骨架。
通过使用Blast2GO与GO数据库的比对,21 777条Unigene获注释信息,在利用WEGO对注释信息进行分类统计,得到136 004个GO功能注释。由分类结果可知:生物学过程最多60 216条,占44.27%,其次是细胞组分,49 219条,占36.19%,最后是分子功能,26 569条,占19.54%。这三大功能分类又可分为51个亚类,其中生物学过程19个亚类,细胞组分15个亚类,分子功能17个亚类。生物学过程中,涉及代谢过程、细胞过程和单一有机体进程的Unigene较多,分别有14 761,12 924,10 871条;细胞组分中涉及较多的是细胞、细胞部分和膜类,分别有10 018,9 976,8 784条;分子功能中涉及较多的有催化活性和结合功能,分别有11 902,10 544条。与其他物种的表达丰度基本一致。具体种类和数量见图3。
将Unigene 与KEGG比对,进行Pathway注释,获得基因产物在细胞的代谢途径以及这些基因产物的功能。比对结果显示有9 648条序列得到9 325个注释,共涉及到237个KEGG标准代谢通路。按基因获得注释量的多少进行排序,选取前10个见表3,涉及碳代谢的Unigene数量最多有392条,占4.20%,其次是与氨基酸的生物合成相关的Unigene,有343条,占3.68%,其余主要富集于核糖体、嘌呤代谢、糖酵解和糖异生等代谢途径。
通过在数据库中查找已有的基因信息和本地Blast比对,共找出黄酮合成相关基因28条,结构基因21条,调节基因7条,见表4。根据苹果[3]中根皮苷的合成途径可知,苯丙氨酸经过苯丙氨酸解氨酶(47968_c1_g1)、肉桂酸羟化酶(46682_c0_g1)、4香豆酰CoA连接酶(42305_c0_g1)的催化,生成香豆酰CoA;乙酰CoA被乙酰CoA羧化酶羧化而成丙二酸单酰CoA。二者经查耳酮合成酶(43222_c0_g1)催化缩合而成查耳酮,紧接着被糖基转移酶(38697_c0_g1)糖基化而成根皮苷。
本研究利用MicroSatallite(MISA)软件找出全部的SSR,总计18 161个,其中单碱基型重复最为丰富,有7 346个,占总量40.45%,在这之中A/T类型分布占其96.31%。其次是双碱基型重复,6 618个,占总量36.44%,其中AG/CT类型分布占其总量78.57%。其他类型依次为:三碱基型重复,3 843个,占21.16%;四碱基型重复,191个,占1.05%;五碱基型重复,66个,占0.36%;最后是六碱基型重复,97个,占0.53%,见图4。通过对多穗柯SSR的研究,将为多穗柯的遗传标记研究提供非常重要物质资源和依据。
3 讨论
近年来,随着多穗柯的甜味和保健作用,尤其是降糖作用被发现,市场需求逐渐变大,研究工作也不断的深入。为更好地探索多穗柯中黄酮类化合物合成,本研究采用RNA-seq技术,获得6 Gb多穗柯转录组数据,经过拼接组装得到41 043条Unigene,N50的长度为1 472 bp,相对于其他已测序的物种,如油松的N50是744 bp[18];芝麻的是1 006 bp[19],组装效果好,完整性高。
通過与7个数据库比对,总共有30 223条Unigene获得注释信息。根据KEGG pathway分析和已知的基因信息,找出28条黄酮合成相关基因,不仅有CHS(查耳酮合成酶)、CHI(查耳酮异构酶)、IFS(异黄酮合成酶)等关键酶基因,还有一些比较重要的基因,如PAL(苯丙氨酸解氨酶)基因、AAC(乙酰辅酶A羧化酶)基因、ANS(花青素苷合成酶)基因,与草麻黄[20]转录组中发现的黄酮合成相关基因大部分一致。通过转录组的组装分析以及黄酮合成相关基因的挖掘,为后续对多穗柯的研究奠定了基础。
[参考文献]
[1] 何春年, 彭勇, 肖伟, 等. 多穗柯甜茶的研究进展[J]. 时珍国医国药, 2012, 23(5): 1253.
[2] 周瑶, 李伟, 曲欣楠, 等. 天然二氢查耳酮类化合物分布及生物活性研究进展[J]. 中国野生植物资源, 2014, 33(6): 35.
[3] Gosch C, Halbwirth H, Stich K. Phloridzin: biosynthesis, distribution and physiological relevance in plants[J]. Phytochemistry, 2010, 71(8/9): 838.
[4] 谭飔, 周志钦. 根皮苷研究进展[J]. 食品与发酵工业, 2013, 39(8):182.
[5] 张召宝, 侯林, 潘晴, 等. 中草药高通量转录组研究进展[J]. 中国中药杂志, 2014, 39(9): 1553.
[6] 吴琼, 孙超, 陈士林, 等. 转录组学在药用植物中的研究应用[J]. 世界科学技术——中医药现代化, 2010, 12(3): 457.
[7] Deng Y Y, Li J Q, Wu S F, et al. Integrated nr database in protein annotation system and its localization[J]. Comput Eng, 2006, 32(5): 71.
[8] Apweiler R, Bairoch A, Wu C H, et al. UniProt: the universal protein knowledge base[J]. Nucleic Acids Res, 2004, 32(1):115.
[9] Ashbuener M, Ball C A, Blake J A, et al. Gene ontology: tool for the unification of biology[J]. Nat Genet, 2000, 25(1): 25.
[10] Koonin E V, Fedorova N D, Jackson J D, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes[J]. Genome Biol, 2004, 5(2): R7.
[11] Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome[J]. Nucleic Acids Res, 2004, 31(1): 277.
[12] Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases[J]. Nucleic Acids Res, 2001, 39(2): 316.
[13] Eddy S R. Profile hidden Markov models[J]. Bioinformatics, 1998, 14(9): 755.
[14] Finn D R, Bateman A, Clements J, et al. Pfam: the protein families database[J]. Nucleic Acids Res, 2014,42 (Database issue):222.
[15] 康亚兰, 裴瑾, 蔡文龙, 等. 药用植物黄酮类化合物代谢合成途径及相关功能基因的研究进展[J]. 中草药, 2014, 45(9): 1336.
[16] 郭欣慰, 黄丛林, 吴忠义, 等. 植物类黄酮生物合成的分子调控[J]. 北方园艺, 2011(4): 204.
[17] 罗文永, 胡骏, 李晓方. 微卫星序列及其应用[J]. 遗传, 2003, 25(5): 615.
[18] Niu S H, Li Z X, Yuan H W, et al. Transcriptome characterisation of Pinus tabuliformis and evolution of genes in the Pinus phyloheny[J]. BMC Genomics, 2013, 14(1): 167.
[19] 魏利斌, 苗紅梅, 张海洋. 芝麻发育转录组分析[J]. 中国农业科学, 2012, 45(7): 1246.
[20] 马婧, 成铁龙, 孙灿岳, 等. 草麻黄高通量转录组分析及黄酮类代谢途径相关基因的鉴定[J]. 浙江农业学报, 2016, 28(4): 609.
