
金属串配合物[CoMCo(dpa)4(NCS)2](M=Co,Ni,Pd,Pt)结构和自旋过滤性质研究
2016-07-13 09:25:15吴子文周沃华丁丹丹罗一帆徐志广
华南师范大学学报(自然科学版) 2016年1期
吴子文, 周沃华, 丁丹丹, 陈 蓉, 许 旋,2,3*, 罗一帆,2,3*, 徐志广,2,3
(1.华南师范大学化学与环境学院,广州 510006; 2.教育部环境理论化学重点实验室,广州 510006;3.广州市能源转化与储能材料重点实验室,广州 510006)
金属串配合物[CoMCo(dpa)4(NCS)2](M=Co,Ni,Pd,Pt)结构和自旋过滤性质研究
吴子文1, 周沃华1, 丁丹丹1, 陈蓉1, 许旋1,2,3*, 罗一帆1,2,3*, 徐志广1,2,3
(1.华南师范大学化学与环境学院,广州 510006; 2.教育部环境理论化学重点实验室,广州 510006;3.广州市能源转化与储能材料重点实验室,广州 510006)
摘要:应用密度泛函理论B3LYP方法对金属串配合物[CoMCo(dpa)4(NCS)2](1:M=Co, 2:M=Ni, 3:M=Pd, 4:M=Pt; dpa=二吡啶胺)的成键性质和自旋过滤效应进行了研究,结果表明:配合物1的基态为二重态,金属链形成三中心三电子σ键σ*0);而配合物2~4的基态均为反铁磁耦合单重态(AF态),对应的最低能量高自旋态(HS态)分别为三重态、七重态和七重态,单电子分布在两端Co原子上,[CoMCo]6+链具有三中心四电子σ键σ*1).配合物1~4均具有自旋过滤效应,电子传输通道主要为β-自旋σnb轨道,与费米能级的距离大小为1<2<3≈4.电场作用下,1~4的高电势端Co2—N4键增长而低电势端Co3—N7键缩短,Co—M平均键长略为缩短,Co—M键增强;电场作用下金属原子的自旋密度和电荷密度变化很小,电磁性质稳定;电场作用下σnb轨道分布仍保持沿金属轴方向离域,LUMO-HOMO能隙减小,有利于电子输运.
关键词:金属串配合物; 密度泛函理论; 自旋过滤; 电场作用; 对称破损
金属串配合物MnL4X2以其独特的结构表现出特殊的电磁性质[1-5],有望作为自旋过滤器分子器件的材料.这类分子的结构特征是金属原子M和轴向配体呈线性排列,桥联配体多吡啶胺L螺旋环绕金属链.目前,很多金属串配合物相继被合成并表征[1,6-12].其中,含3个金属原子且以NCS-为轴向配体的金属串配合物因S原子可与金电极直接相连构成分子器件而备受关注.自2004年,PENG研究组制备了一系列含NCS-轴向配体的Co、Ni、Cr三、五核金属串配合物[13-14],通过扫描隧道显微镜(STM)观察发现Co3(dpa)4(NCS)2具有较好的导电性,dpa为二吡啶胺(dipyridylamine).磁性实验结果表明Co3(dpa)4(NCS)2和Co3(dpa)4Cl2的基态均为二重态.GEORGIEV等[15-16]用密度泛函理论(DFT)结合非平衡态格林函数(NEGF)方法研究Co3(dpa)4(NCS)2发现:电子主要通过靠近费米能级的β-自旋单占σnb轨道传输,且存在90%以上的自旋过滤效应(SFE).因金属价电子结构对金属串配合物的结构和性质影响很大,为调节电磁性质,LIU等[11]合成了CoPdCo(dpa)4Cl2,经实验和DFT计算均表明,其基态为反铁磁单重态(AF态),端基Co采取自旋量子数S=3/2的高自旋态(HS),中间Pd为抗磁性.TABOOKHT等[17]用DFT方法研究了含22个价电子的CoMCo(dpa)4X2(M=Pd, Ni; X=Cl-, NCS-),发现配合物的基态均为AF态.M为Pd时,AF对应HS为七重态;而M为Ni时,AF对应的HS为三重态.但[CoMCo(dpa)4X2](M=Ni, Pd, Pt)配合物是否具有自旋过滤效应未见研究报道.
分子器件需在外电场环境下工作,要设计分子器件须了解电场对其结构的影响.我们对金属串配合物[Ru3(dpa)4]L2(L=Cl, C≡N, C≡CPh)[18]、[Cu2M(npa)4Cl]+(M=Pt,Pd,Ni)[19]和M3(dpa)4Cl2(M=Co, Rh, Ir)[20]结构与外电场关系的理论研究发现,不同价电子数的金属串配合物的几何结构和电子结构受电场影响差异较大,对CoMCo(dpa)4(NCS)2的结构受电场的影响未见报道.本文以含21个价电子金属的Co3(dpa)4(NCS)2(1)为参照,用DFT方法研究含22个价电子的CoMCo(dpa)4(NCS)2(2:M=Ni, 3:M=Pd, 4:M=Pt)的电子组态、自旋过滤效应、M—M相互作用以及电场作用下几何结构和电子结构的变化规律,为设计该类分子器件提供理论参考.
1计算方法
以1和CoPdCo(dpa)4Cl2的晶体结构为初始结构,并据此设计2~4的初始结构,均为D4点群.采用B3LYP和BP86这2种泛函对配合物1~4的各种可能多重态进行几何优化,对C、N和H原子采用6-31G*基组,对Cl和S原子采用6-311G*基组,对金属原子采用LanL2DZ基组.对AF态使用对称性破损(BS)方法[21]计算.对优化构型进行振动频率分析未发现虚频,并利用自然键轨道理论(NBO)[22]分析.从表1可知,2种泛函计算的配合物1的基态(标为HS态)均为二重态,与磁性实验[8]1270一致;而B3LYP和BP86方法计算的CoPdCo(dpa)4Cl2的最低能量HS态则分别为七重态和单重态,只有B3LYP的计算结果与LIU等的实验和理论结果[11]9603-9604一致.因此选择分析B3LYP泛函对配合物1~4的计算结果.结果表明配合物2的最低能量HS态为三重态,3和4的都为七重态.
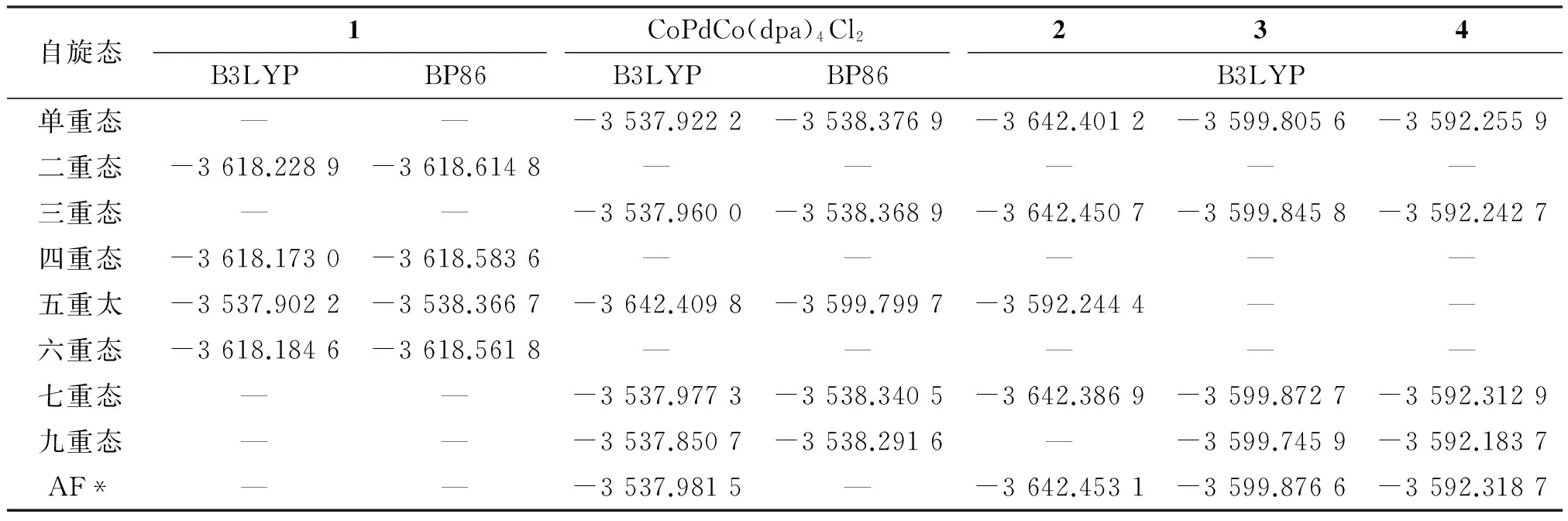
表1 配合物1~4和CoPdCo(dpa)4Cl2不同自旋态的分子能量
因金属串配合物的电子传输方向为金属轴(z轴),为模拟分子器件的工作环境,固定金属轴两端S原子的坐标,沿z轴负方向(图1)施加均匀电场,应用B3LYP方法对1~4的HS态以及2~4的AF态在电场作用下进行几何优化,电场范围为0.000~2.571 V/nm.采用Multiwfn软件[23]模拟态密度(DOS)图,其它计算均应用Gaussian 09[24]软件包.

图1 配合物1的结构及电场方向
2结果与讨论
2.1零电场下配合物的结构与性质
2.1.1配合物的几何结构1的二重态HS和CoPdCo(dpa)4Cl2的AF态的几何参数计算值与文献[8]1266、[11]3603的实验值接近(表2),表明所选计算方法合适.2~4的最低能量高自旋态(HS)与相应基态(AF)的几何参数和变化规律接近.比较2与1的结果可知,d8电子的Ni2+取代d7电子的Co2+后,Co—Ni、Co2—N4(轴向)和Co2—N2键均增长,Ni—N1键较1中Co1—N1键短.随M的周期数增大,M2+原子半径增大,Co—M、Co2—N2和M—N1键长逐渐增长,Co2—N4键长逐渐缩短.AF态中,对于金属链方向,3的Co2—N4和Co—Pd键长分别为0.201 3 nm和0.256 5 nm,均比CoPdCo(dpa)4Cl2的Co2—Cl4和Co—Pd键短;但关于桥联配体方向,3中的Co2—N2和Pd—N1键则比后者的长.配合物1的基态和2~4的AF态Co—M键的Wiberg键级则依次为0.281 8、0.201 8、0.222 2、0.2772,Co2—N4键级依次为0.412 6、0.389 4、0.445 0、0.445 9.表明d8电子的M2+取代Co2+后,使Co—M、Co2—N4相互作用减弱,但随着M的周期数增大,Co—M、Co2—N4相互作用增强.
2.1.2零电场下配合物的自旋密度表3为零电场下配合物的自旋密度.HS态下,配合物1中3个Co原子均有单占电子,两端的Co原子单电子自旋向上,中间的Co自旋向下,呈反铁磁耦合.d8电子M2+取代中间的Co2+后,配合物2~4中间金属原子的自旋密度几乎为0,呈抗磁性,自旋密度主要集中在两端的磁中心Co(Ⅱ)原子上呈铁磁耦合.与HS态不同的是,AF态中集中于两端的磁中心Co(Ⅱ)原子的自旋电子呈反铁磁耦合.2~4两种状态下的自旋密度分布情况与Ni3(dpa)4Cl2的分布情况[25]相似,只是自旋密度略向轴向配体N4离域,有利于耦合作用.
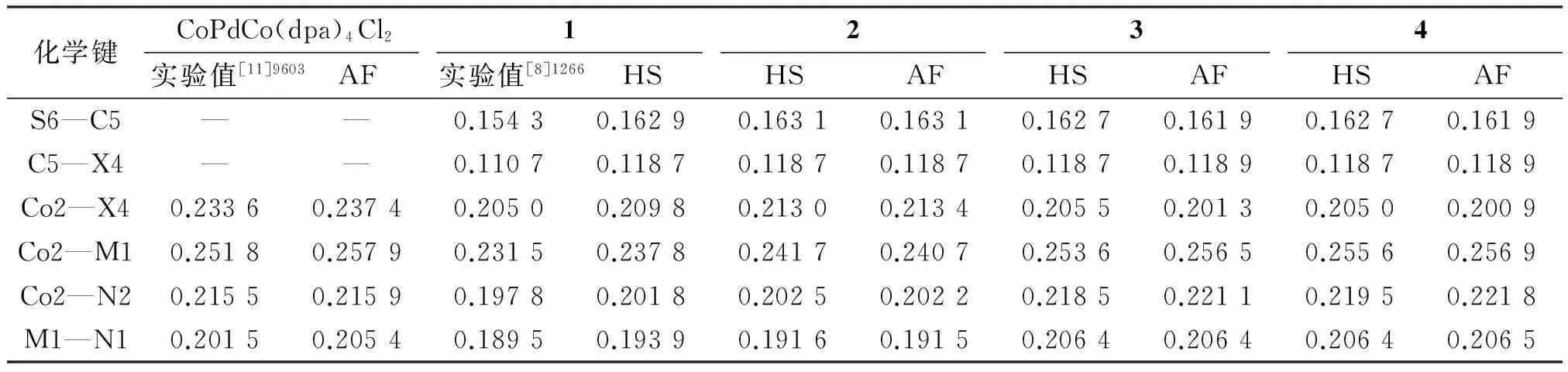
表2 配合物1~4优化的部分键长
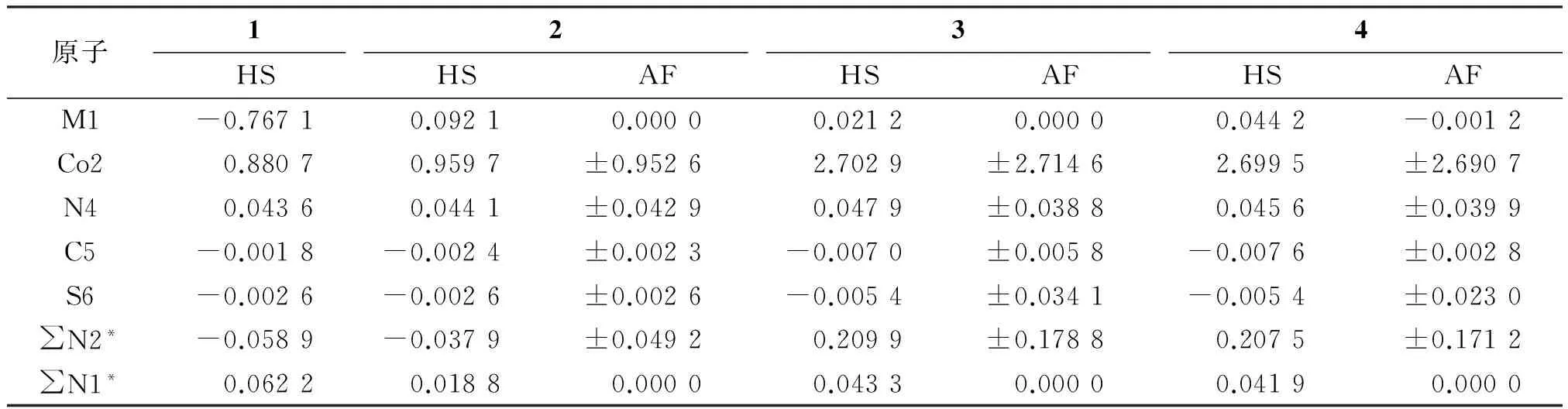
表3 配合物1~4的自旋密度
*∑N1和∑N2是N原子的总自旋密度.

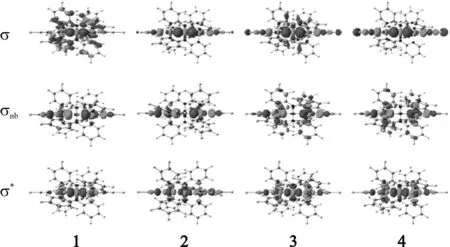
图2 配合物1~4的HS态分子轨道图
Figure 2Molecular orbitals diagrams for HS states of complexes 1~4

图3 配合物1~4的HS态金属特征分子轨道能级图
自旋过滤[26]是指自旋取向不同的隧穿电子通过隧道结的势垒时,由于势垒层具有铁磁性,在居里温度下其导带发生交换劈裂,自旋向上电子遇到的势垒较低,而自旋向下电子遇到的势垒则较高,通过势垒后电子产生自旋极化.自旋极化强度可用公式P=(Nβ-Nα)/(Nα+Nβ)表示,其中Nα和Nβ分别为费米能级附近α、β自旋电子的态密度.自旋极化强度可衡量SFE.
对于单占的σnb轨道,配合物1的α-自旋σnb轨道位于费米能级以下4.907 eV处,而β-自旋σnb轨道为LUMO轨道,刚好位于费米能级上,β-自旋的σnb轨道被认为是电子传输的主要通道[15]5595.由图4可知:电子传输通道β-自旋σnb在费米能级处(虚线)有1个分波态密度(PDOS)峰,电子较活跃,而α-自旋的PDOS峰远离费米能级,费米能级附近σnb的α、β电子态密度分布不对称,证实了关于1存在SFE的观点[15]5595.
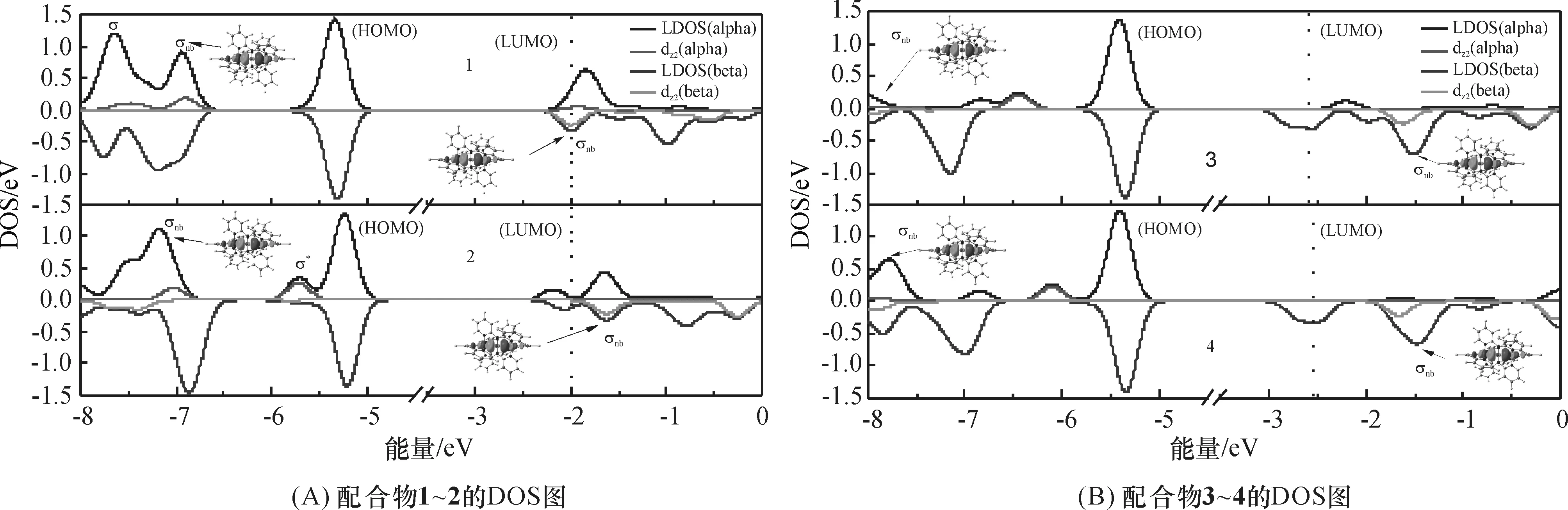
图4 配合物1~4的HS态DOS图
而配合物2~4中,β-自旋的σnb轨道比费米能级分别高0.436 7、1.179和1.032 eV,α-自旋的σnb轨道比费米能级低4.956、5.402和5.508 eV,在费米能级附近σnb的α、β电子态密度分布不对称,故推测配合物2~4也具有较高的SFE,但传输通道σnb与费米能级距离为4≈3>2>1.
2.2外电场下配合物的性质
2.2.1外电场对配合物NPA电荷的影响配合物1~4中两端Co的自然电荷布居(NPA)正电荷都大于中间的金属原子M(图5,图中所注数值为零电场时的NPA电荷),当d8电子的Ni取代d7电子的Co后,桥联配体N原子与两端Co的作用减弱,Co2—N2键增长.随着M的周期数增大,Co2—N2键削弱更显著,故桥联配体的电子向两端Co的转移减弱,Co的正电性增大.电场作用下两端S的电荷密度变化最明显(图5),电场作用使低电势端S9的负电荷减小,而高电势端S6的负电荷增大,负电荷从低电势端向高电势端转移,符合物理学规律.而金属原子、桥联配体上的电荷在电场作用下基本没有变化,这支持了关于dpa-配体对[M3(dpa)4(SCN)2] (M=Cr, Co, Ni)的电荷输运起屏蔽作用的观点[27]3642.配合物2~4的AF态电荷密度在电场作用下的变化情况与HS态基本一致.
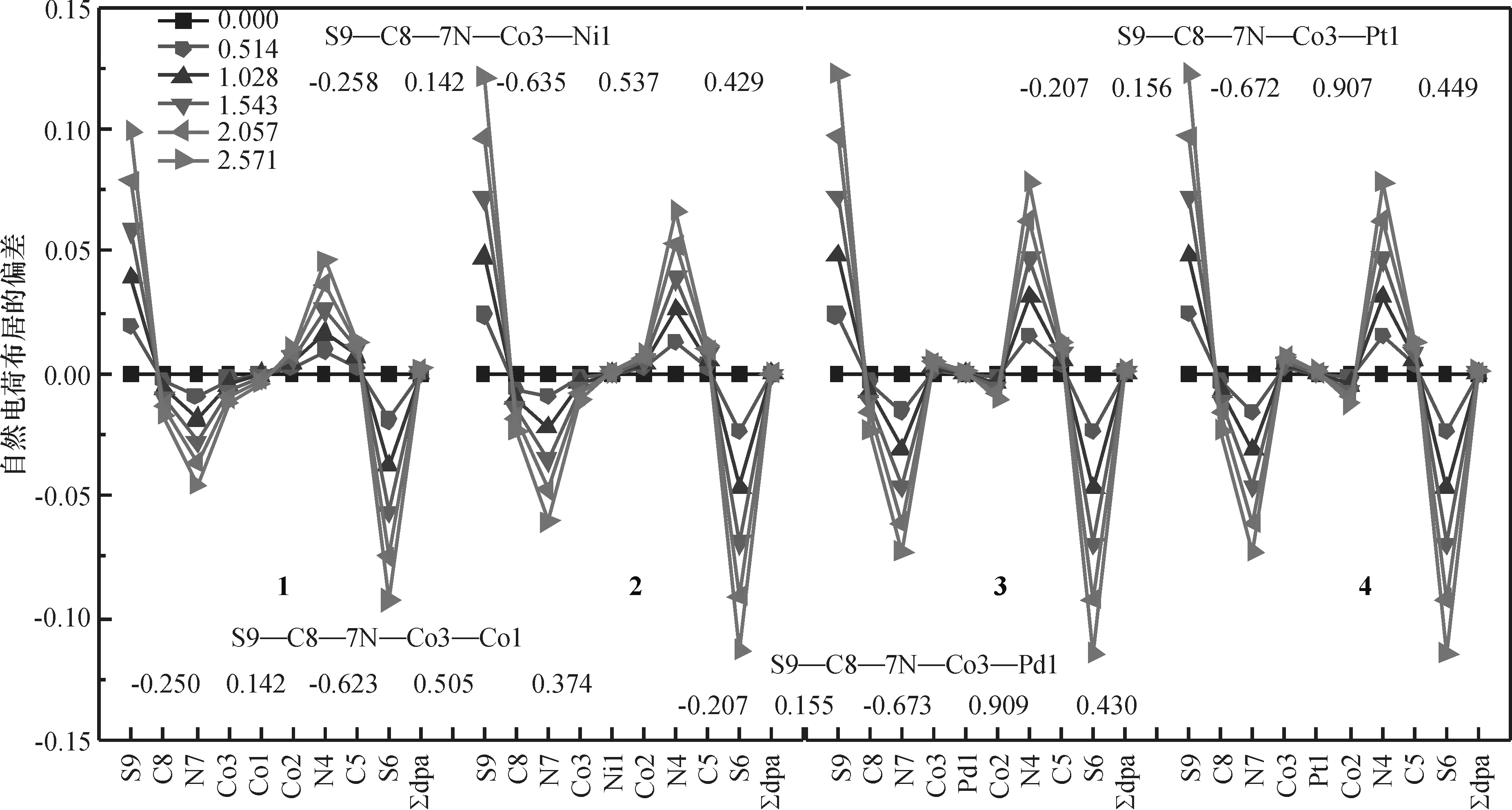
图5 电场作用下配合物1~4的HS态自然电荷布居的变化
2.2.2外电场对配合物几何构型的影响在-z方向电场作用下,Co—M、轴向Co—N键长都发生了规律性变化,轴向Co—N键长受电场的影响较大.AF态的键长变化情况与HS态基本一致.电场作用下键长的变化主要受带电粒子的移动、化学键的束缚、极化和离域等因素影响.由于Co—M、Co—N键较弱,且金属原子的正电性较大,因此,带电粒子的移动是电场作用下键长变化的主要因素.在电场作用下(图6),带正电的Co2、Co3沿电场方向移动,因此Co2—N4键增长而Co3—N7键缩短,与Co3(dpa)4Cl2在电场作用下的键长变化情况[20]1228一致.当电场达到2.571 V/nm时,Co2—N4键分别增长0.005 55、0.005 44、0.003 97和0.003 94 nm,Co3—N7键则分别缩短0.005 04、0.005 01、0.003 70和0.003 73 nm,随着M的周期数增大,Co—N键增强,Co—N键长随电场变化减小,且Co2—N4键的增长比Co3—N7键的缩短显著,使Co—M平均键长略微减小,故Co—M相互作用稍为增强.由于两端Co的正电荷比中间M的高,故两端Co向低电势端(-z方向)的移动比M更显著,使2~4中Co2—M1键减短,Co3—M键增长,且随着两端Co的正电荷增大,Co—M键长的变化更为显著,故Co—M键长的变化显著性次序为4>3>2>1.
2.2.3外电场对配合物自旋密度的影响分析外电场对电子自旋密度的影响有助于对分子器件磁性的理解.均匀外电场使HS态自旋密度发生了规律性变化(图7,图中数值为零电场时的自旋密度),均匀外电场使配合物1~2的HS态低电势端Co3的自旋密度减小而高电势端Co2的的自旋密度增大,配合物3、4则与之相反,但四者变化均很小,但变化很小.当电场达到2.571 V/nm时,变化最大的配合物1中Co3的自旋密度变化值仅0.034 78,而2~4中因自旋密度局域在两端的Co上,故各原子的自旋密度在电场下的变化更小.4个dpa-配体的自旋密度之和∑dpa-基本不变,表明电场作用下桥联配体与金属链和轴向配体间没有自旋密度的转移.2~4的AF态自旋密度(图8)在电场中的变化规律与HS态相似,受电场的影响也很小.因此,不管是AF态还是HS态,电场作用下化合物的磁性稳定.

图6 电场作用下配合物1~4的HS态和2~4的AF态部分键长的变化
2.2.4外电场对前线分子轨道的影响前线分子轨道是决定分子多种物理化学性质的关键因素,也影响着分子器件的电子输运性质,分析前线分子轨道能级、空间分布以及最低空轨道LUMO与最高占据轨道HOMO的能隙(Eg)随电场的变化有助于了解分子的电子传输性质.
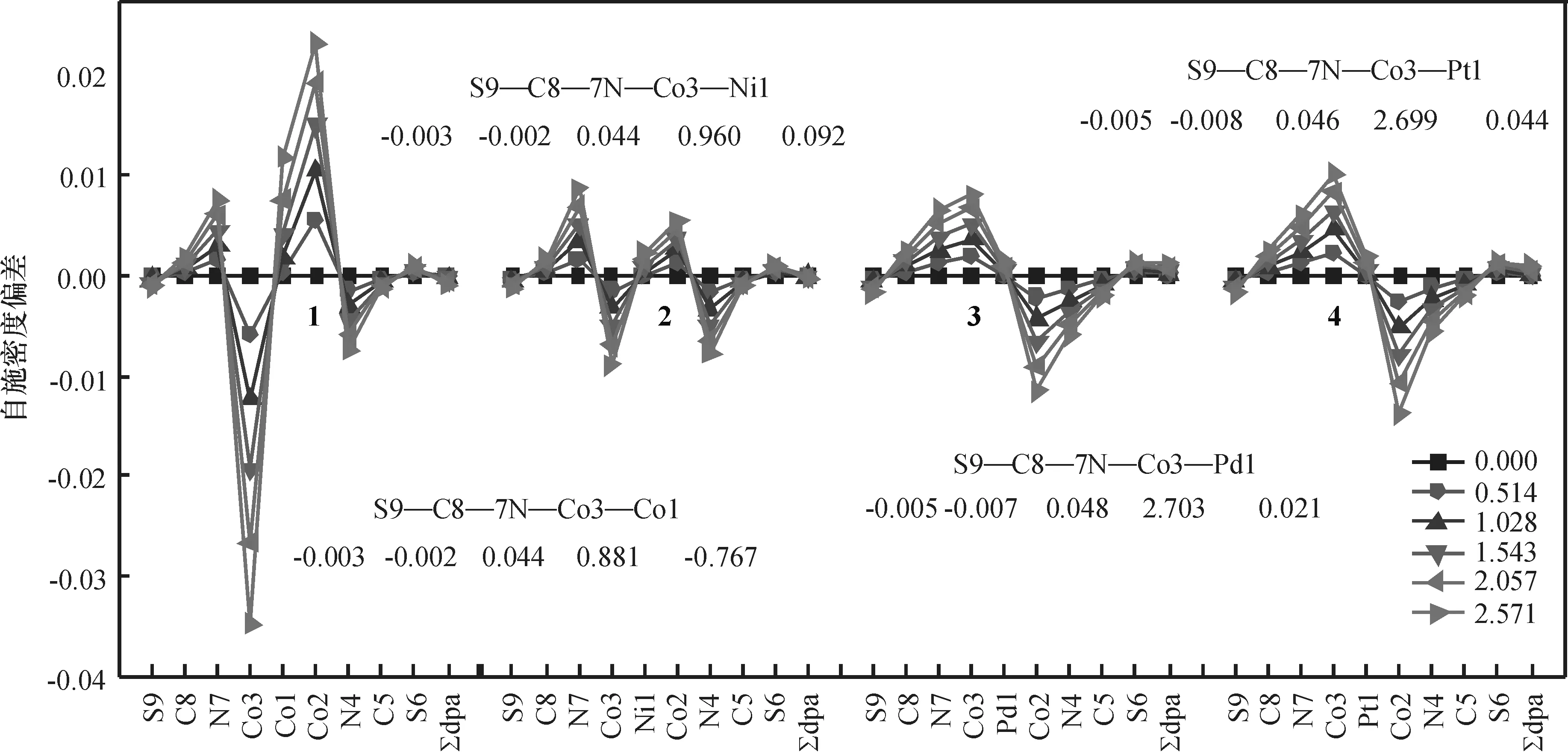
图7电场作用下配合物1~4的HS态自旋密度的变化
Figure 7Electric field dependence of spin densities for HS states of complexes 1~4
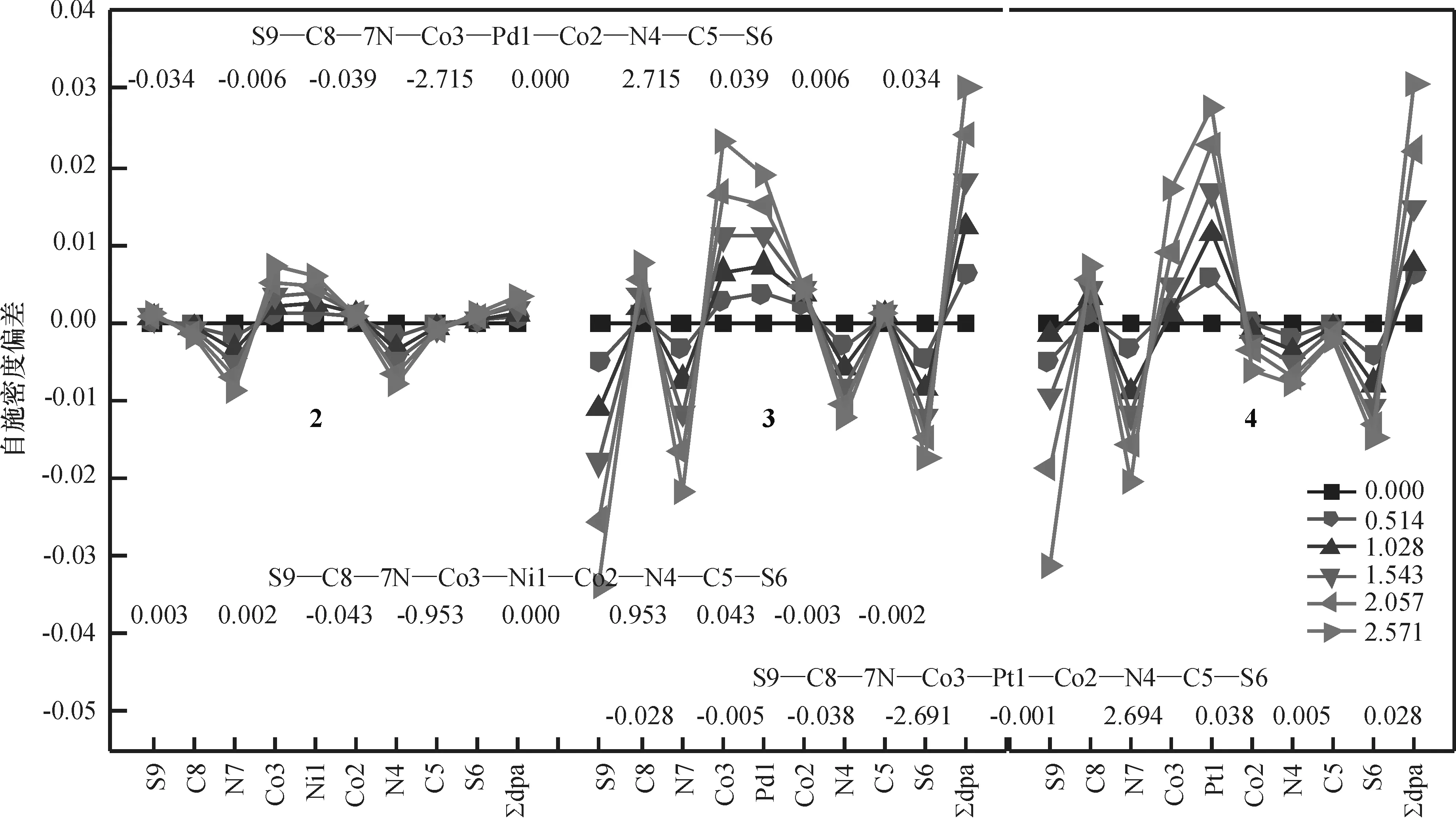
图8 电场作用下配合物1~4的AF态自旋密度的变化
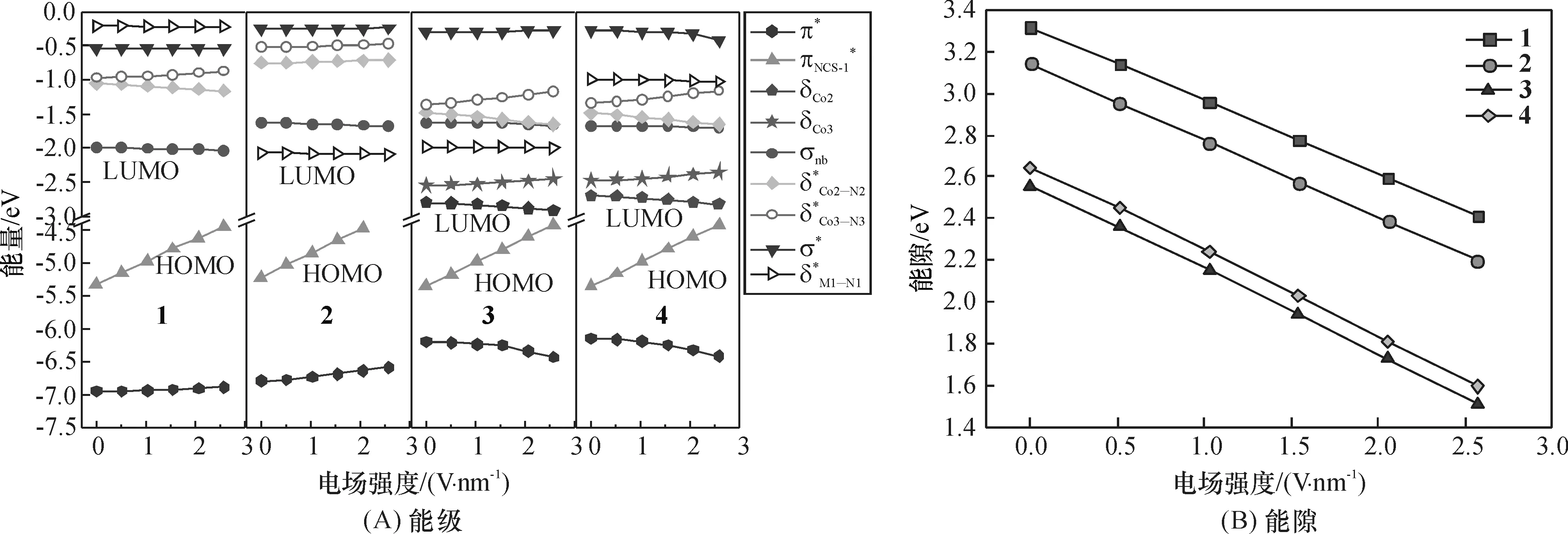
图9 电场作用下配合物1~4的HS态β-自旋分子轨道能级和能隙的变化
表4电场作用下配合物2分子HS态β自旋分子轨道空间分布的变化
Table 4Spatial distribution of β-spin molecular orbitals for HS states of complexes 2 under electric field

3结论

参考文献:
[1]WU L P, FIELD P, MORRISSEY T, et al. Crystal structure and electronic properties of dibromo- and dichloro-tetrakis [μ3-bis(2-pyridyl)amido] tricopper (II) hydrate[J]. Dalton Transactions, 1990, 12:3835-3840.
[2]YANG E C, CHENG M C, TSAI M S, et al. Structure of a linear unsymmetrical trinuclear cobalt(II) complex with a localized CoII-CoII bond: dichlorotetrakis[μ3-bis(2-pyridyl)amido] tricobalt(II)[J]. Chemical Communications, 1994, 20:2377-2378.
[3]CLÉRAC R, COTTON F A, DANIELS L M, et al. Tuning the metal-metal bonds in the linear tricobalt compound Co3(dpa)4Cl2: bond-stretch and spin-state isomers[J]. Inorganic Chemistry,2001, 40(6):1256-1264.
[4]BERRY J F, COTTON F A, MURILLO C A, et al. An efficient synthesis of acetylide/trimetal/acetylide molecular wires[J]. Inorganic Chemistry, 2004, 43(7):2277-2283
[5]HSU L Y, HUANG Q R, JIN B Y. Charge transport through a single molecular wire based on linear multimetal complexes: a non-equilibrium Green’s Function approach[J]. Journal of Physical Chemistry C, 2008, 112(28):10538-10541.
[6]ADULDECHA S, HATHAWANY B. Crystal structure and electronic properties of tetrakis [μ3-bis (2-pyridyl)amido] dichlorotrinickel(II)-water-acetone (I/0.23/0.5)[J]. Dalton Transactions, 1991, 4:993-998.
[7]COTTON F A, DANIELS L M, JORDAN G T I V. Efficient preparation of a linear, symmetrical, metal-metal bonded tricobalt compound: should we believe there is a bond stretch isomer?[J]. Chemical Communications, 1997, 5:421-422.
[8]CLÉRAC R, COTTON F A, JEFFERY S P, et al. Compounds with symmetrical tricobalt chains wrapped by dipyridylamide ligands and cyanide or isothiocyanate ions as terminal Ligands[J]. Inorganic Chemistry, 2001, 40(6):1265-1270.
[9]BERRY J F, COTTON F A, FEWOX C S, et al. Extended metal atom chains (EMACs) of five chromium or cobalt atoms: symmetrical or unsymmetrical?[J]. Dalton Transactions, 2004, 15:2297-2302.
[10]ROHMER M M, LIU I P C, LIN J C, et al. Structural, magnetic, and theoretical characterization of a heterometallic polypyridylamide complex[J]. Angewandte Chemie International Edition, 2007, 46(19):3533-3536.
[11]LIU I P C, LEE G H, PENG S M, et al. Cu-Pd-Cu and Cu-Pt-Cu linear frameworks: synthesis, magnetic properties, and theoretical analysis of two mixed-metal complexes of dipyridylamide(dpa), isostructural, and isoelectronic with [Cu3(dpa)4Cl2]+[J]. Inorganic Chemistry, 2007, 46(23):9602-9608.
[12]KUO J H, TSAO T B, LEE G H, et al. An extended metal chain with the 2,7-Bis(dipyridyldiamino)-1,8-naphthyridine (H4bdpdany) ligand -The longest even-numbered metal chain complex[J]. European Journal of Inorganic Chemistry, 2011, 13:2025-2028.
[13]LIN S Y, CHEN I W P, CHEN C H, et al. Effect of metal-metal interactions on electron transfer: an STM study of one-dimensional metal string complexes[J]. Journal of Physical Chemistry B, 2004, 108(3):959-964.
[14]CHEN I W P, FU M D, TSENG W H, et al. Conductance and stochastic switching of ligand-supported linear chains of metal atoms[J]. Angewandte Chemie, 2006, 118(35):5946-5950.
[15]GEORGIEV V P, MCGRADY J E. Efficient spin filtering through cobalt-based extended metal atom chains[J]. Inorganic Chemistry, 2010, 49(12):5591-5597.
[16]GEORGIEV V P, SAMEERA W M C, MCGRADY J E. Attenuation of conductance in cobalt extended metal atom chains[J]. Journal of Physical Chemistry C, 2012, 116(38):20163-20172.
[17]TABOOKHT Z, DE G C, LPEZ X. Towards a low-spin configuration in extended metal atom chains. Theoretical study of trimetallic systems with 22 metal electrons[J]. Dalton Transactions, 2012, 41:498-504.
[18]许旋, 张胜楠, 莫小婵, 等. 电场作用下金属串配合物[Ru3(dpa)4]L2(L=Cl, C≡N, C≡CPh)结构的理论研究[J]. 华南师范大学学报:自然科学版, 2015, 47(2):39-47.
XU X, ZHANG S N, MO X C, et al. Theoretical study on structures of metal string complexes[Ru3(dpa)4]L2(L=Cl, C≡N, C≡CPh)under the electric field[J]. Journal of South China Normal University: Natural Science Edition, 2015, 47(2):39-47.
[19]黄晓, 谭莹, 许旋, 等. 电场对杂金属串配合物[CuCuM(npa)4Cl]+(M=Pt, Pd, Ni)结构影响的理论研究[J].化学学报, 2012, 70(18):1979-1986.
HUANG X, TAN Y, XU X, et al. Theoretical studies on structures of heterometal string complexes[CuCuM(npa)4Cl]+(M=Pt, Pd, Ni) under the electric field[J]. Acta Chimica Sinica, 2012, 70(18):1979-1986.
[20]黄燕, 黄晓, 许旋, 等. 电场对M3(dpa)4Cl2(M=Co, Rh, Ir)金属串配合物结构影响的理论研究[J].物理化学学报,2013, 29(6):1225-1232.
HUANG Y, HUANG X, XU X, et al. Effects of electric field on the structures of metal string complexes M3(dpa)4Cl2(M=Co, Rh, Ir; dpa=dipyridylamide)[J].Acta Physico-Chimica Sinica, 2013, 29(6):1225-1232.
[21]NOODLMAN L. Valence bond description of antiferromagnetic coupling in transition metal dimers[J]. Journal of Chemical Physics, 1981, 74(10):5737-5743.
[22]GLEDENING E D, REED A E, CARPENTER J E. Gaussian 03, Version D.01, NBO Version 3.1[CP].Gaussian Inc., Wallingford CT, 2003.
[23]LU T, CHEN F. Multiwfn: a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5):580-592.
[24]FRISCH M, TRUEKS G, SCHLEGEL H B, et al. Gaussian 09, Revision B.01[CP]. Gaussian Inc., Pittsburgh, PA, 2009.
[25]YASUTAKA K, TORU M, YASUYUKI N, et al. Theoretical studies of electronic structures, magnetic properties and electron conductivities of one-dimensional Nin(n=3, 5, 7) complexes[J]. Dalton Transactions, 2013, 42(45):16203.
[26]MOODERA J S, HAO X, GIBSON G A, et al. Electron-spin polarization in tunnel junctions in zero applied field with ferromagnetic EuS barriers[J]. Physical Review Letters, 1988, 61(5):638.
[27]TSAI T W, HUANG Q R, PENG S M, et al. Smallest electrical wire based on extended metal-atom chains[J]. Journal of Physical Chemistry C, 2010, 114(8):3642.
【中文责编:谭春林英文责编:李海航】
Theoretical Studies on the Structures and Spin Filtering Property of Metal String Complexes [CoMCo(dpa)4(NCS)2](M=Co,Ni,Pd,Pt)
WU Ziwen1, ZHOU Wohua1, DING Dandan1, CHEN Rong1, XU Xuan1,2,3*, LUO Yifan1,2,3*, XU Zhiguang1,2,3
(1.School of Chemistry & Environment, South China Normal University, Guangzhou 510006;2. Key Laboratory of Theoretical Chemistry of Environment, Ministry of Education, Guangzhou 510006;3.Key Laboratory of Materials for Energy Conversion and Storage of Guangzhou, Guangzhou 510006)
Abstract:The bonding and spin filtering properties of metal string complexes [CoMCo(dpa)4(NCS)2](M=Co, Ni, Pd, Pt; dpa=dipyridylamine) have been investigated with the density functional theory B3LYP method. The results show that the ground state of complex 1 is doublet, and there is a 3-center-3-electron σ bondσ*0) delocalized over thechain. However, the ground states of complexes 2~4 are antiferromagnetic(AF) singlet, corresponded to the high spin configurations with two unpaired, six unpaired and six unpaired electrons, respectively. The unpaired electrons of complexes 2~4 largely localize on the terminal CoIIions and there is a 3-center-4-electron σ bondσ*1) delocalized over the [CoMCo]6+chain. The molecular orbital energy level diagrams and PDOS diagrams show that the spin-β components of singly σnborbital are the dominant transport channel. Complexes 1~4 all possess spin filtering effect. The distance between spin-β components of σnborbital and Fermi levels is 1<2<3≈4. Under the electric field, the Co2—N4 bond lengths at the high potential side increase, while the Co3—N7 distances at the low potential side tend are shortened. The average Co—M distances, the spin densities and NPA charge of metal atoms are less affected by electric field, indicating the electromagnetic properties are stable. Under the electric field, the σnborbital still keeps delocalized along the metal chain and the LUMO-HOMO gaps decrease. This is beneficial for electron transfer.
Key words:metal string complex; density functional theory; spin filtering; electric field effect; broken symmetry
收稿日期:2015-05-13《华南师范大学学报(自然科学版)》网址:http://journal.scnu.edu.cn/n
基金项目:广东省自然科学基金项目(S2012010008763);广东省教育部产学研项目(2010B090400184);广州市科技攻关项目(2011J4300063)
*通讯作者:许旋,教授,Email:xux@scnu.edu.cn;罗一帆,教授,Email:luoyf2004@126.com.
中图分类号:O641
文献标志码:A
文章编号:1000-5463(2016)01-0058-09
