
IL-33及其受体ST2在D-GalN/LPS诱导的急性肝功能衰竭小鼠中的表达及意义
2016-05-23 00:44姜绍文林兰意项晓刚卢捷王芃莫瑞东刘昱含蔡伟王晖谢青
肝脏 2016年4期
姜绍文 林兰意 项晓刚 卢捷 王芃 莫瑞东 刘昱含 蔡伟 王晖 谢青
200025 上海交通大学医学院附属瑞金医院感染科
·论著·
IL-33及其受体ST2在D-GalN/LPS诱导的急性肝功能衰竭小鼠中的表达及意义
姜绍文林兰意项晓刚卢捷王芃莫瑞东刘昱含蔡伟王晖谢青
200025上海交通大学医学院附属瑞金医院感染科
【摘要】目的研究D-GalN/LPS诱导的急性肝衰竭小鼠中IL-33及其受体ST2的表达及意义。方法腹腔注射D-GalN(900 mg/kg)/LPS(10 μg/kg)诱导急性肝衰竭小鼠模型。通过q-PCR、Western印迹、ELISA、免疫组织化学染色等实验技术检测IL-33及其受体ST2在不同时间点的动态变化。结果急性肝衰竭小鼠肝内的IL-33 mRNA水平随着肝损伤加重不断增高,肝衰竭时上升至峰值,D-GalN/LPS诱导后7 h,肝组织表现为明显坏死。而肝内ST2L受体蛋白含量在D-GalN/LPS诱导后3 h,未出现明显的肝细胞损伤前已显著升高,之后不断下降,到7 h肝衰竭时其水平降至最低。此外,外周血清中IL-33蛋白水平亦随时间持续升高,在7 h肝衰竭时达高峰,与IL-33 mRNA的动态变化相一致。然而血清sST2蛋白水平在0 h和3 h肝细胞损伤的早期无明显差异,但在5 h肝细胞损伤的中期却显著升高,之后又显著降低。免疫组织化学染色显示急性肝衰竭小鼠肝内IL-33来源于血管内皮细胞和肝血窦细胞核内。结论IL-33及其受体ST2随时间的动态变化与急性肝衰竭的病情进展存在紧密联系,提示IL-33/ST2轴参与了急性肝衰竭的发生发展过程。
【关键词】IL-33;ST2L;sST2;急性肝功能衰竭;动态表达
急性肝功能衰竭是临床常见的严重肝病症候群,进展迅速,病死率高达60%以上,严重威胁人类健康[1]。引起急性肝衰竭最常见的原因包括对乙酰氨基酚过量、特异性药物反应和肝炎病毒感染[2]。目前急性肝衰竭唯一有效的治疗手段仍然是肝移植,但肝源稀缺、费用过高等因素极大地限制了其在临床上的广泛应用。因此,及早地阐明急性肝衰竭的发生发展机制,找到针对性的治疗靶点,有效地阻止病情进展恶化,对于挽救患者生命而言至关重要。既往研究已证实,全身性炎性反应在急性肝衰竭的发病过程中发挥举足轻重的作用。TNF-α、IL-1β、IL-6和IL-18等促炎因子持续造成肝细胞损伤,引发炎性级联反应,最终导致多器官功能衰竭[3, 4]。
IL-33 (Interleukin-33)是IL-1家族的第11个新成员,又称IL-1F11[5]。不但可作为核内转录因子参与基因的表达调控,还可作为细胞因子被分泌到胞外,激活靶细胞,启动免疫反应[6]。研究表明,IL-33参与多种肝脏疾病的发生过程,包括病毒性肝炎、肝脏纤维化和肝细胞肝癌[7-9]。然而,关于IL-33/ST2轴在急性肝衰竭中作用的相关研究较少。本研究通过观察急性肝衰竭小鼠模型中IL-33及其受体ST2的动态表达,探究IL-33/ST2轴是否参与急性肝衰竭的发生发展过程,为其治疗寻找新的潜在靶点。
资料和方法
一、动物和试剂
Balb/C清洁级小鼠,雄性,6~7周龄,体质量20~25 g,购自中国科学院上海实验动物中心,在上海交通大学附属瑞金医院动物房清洁级环境中适应1周后开始试验。试验前12 h禁食不禁水,给药后正常进食进水。
D-GalN和LPS购自美国Sigma公司,TRIzol试剂购自美国Invitrogen公司,反转录试剂盒和SYBR Green Taq试剂盒购自日本Takara公司,IL-33和ST2的ELISA试剂盒以及一抗购自美国R&D公司。
二、急性肝衰竭小鼠模型的建立
分别按照D-GalN 900、500、400、300 mg/kg和LPS 10 μg/kg的药物浓度,给予小鼠腹腔注射,给药后0、3、5、7 h分别处死6只小鼠,留取血清和肝组织。血清存于-80 ℃,一部分肝组织在液氮速冻后存于-80 ℃冰箱,另一部分肝组织甲醛固定。
三、RNA抽提、反转录和q-PCR
用TRIzol试剂提取肝组织总RNA,具体操作按照说明书进行;测定总RNA浓度,配制成等浓度RNA溶液。用反转录试剂盒和SYBR Green Taq试剂盒完成反转录和q-PCR试验,具体操作按照试剂盒说明书进行。IL-33引物由铂尚公司合成,上游引物为5′-ATTTCCCCGGCAAAGTTCAG-3′,下游引物为5′-AACGGAGTCTCATGCAGTAGA-3′。
四、免疫印迹试验
肝组织匀浆后离心、取上清,经BCA法测定蛋白浓度,制备蛋白样本,使相同体积溶液内含有相同质量的蛋白。配制10%的胶,完成蛋白样品和marker上样,电泳。再经过350 mA电流 70 min,将蛋白转移至PVDF膜。在含5%脱脂奶粉的TBST溶液中常温封闭1 h。TBST溶液漂洗,5 min × 3次。ST2一抗(1∶2 000)4℃ 孵育过夜。TBST溶液漂洗同前,抗山羊二抗(1∶5 000,碧云天)常温孵育1 h。TBST溶液漂洗同前,ECL显影液显影,曝光拍照。
五、酶联免疫吸附试验
血清原液检测IL-33,血清1∶20稀释检测ST2,具体操作严格按照R&D试剂盒说明书进行。
六、免疫组织化学染色
5 μm厚的连续石蜡切片常规脱蜡至水,柠檬酸-EDTA修复液98 ℃抗原修复30 min,冷却至室温,TBST缓冲液浸洗10 min × 3次,3%过氧化氢室温10 min灭活内源性过氧化物酶,TBST溶液浸洗同前,0.2%曲拉通室温5 min,TBST溶液浸洗同前,二抗同源血清封闭30 min,IL-33一抗(1∶100)4 ℃ 孵育过夜,TBST溶液浸洗同前,二抗(1∶200)37 ℃孵育30 min,TBST溶液浸洗同前,DAB显色,苏木素复染、分色,冲水返蓝,梯度脱水、透明,中性树脂封片。阴性对照以PBS缓冲液代替一抗。
结果
一、D-GalN 900 mg/kg 联合LPS 10 μg/kg腹腔注射建立急性肝衰竭小鼠模型
不同剂量组合的D-GalN/LPS腹腔注射后小鼠的24 h生存曲线见图1a。D-GalN 900 mg/kg 联合LPS 10 μg/kg可以诱导高死亡率的小鼠急性肝衰竭,与临床急性肝衰竭情况较为接近。D-GalN 900 mg/kg 联合LPS 10 μg/kg腹腔注射后,分别在0、3、5、7 h时间点动态观察小鼠肝脏的转氨酶、肉眼观和HE染色病理改变。随着肝损伤的加重,ALT和AST逐渐升高,至7 h达高峰(图1b)。从0 h到7 h,小鼠肝脏的体积因淤血逐渐增大,色泽由粉红色渐变为暗红色,5 h肝脏表面出现散在点状出血点,7 h肝脏表面满布颗粒状出血点(图1c)。肝组织病理HE染色显示,3 h肝小叶结构健全,部分肝细胞肿胀或出现空泡变性;5 h部分肝细胞发生凋亡,并出现组织内炎性细胞浸润及显著的点状、小片状坏死,肝血窦内开始出现淤血;7 h肝小叶结构完全丧失,肝细胞大块坏死,汇管区大量炎性细胞浸润,肝血窦内充斥血细胞或闭塞不通,微循环障碍严重(图1D-G)。
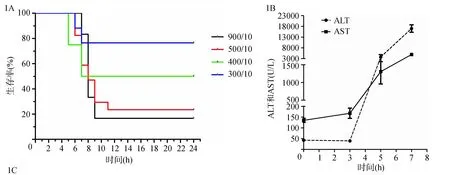
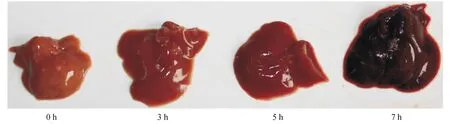
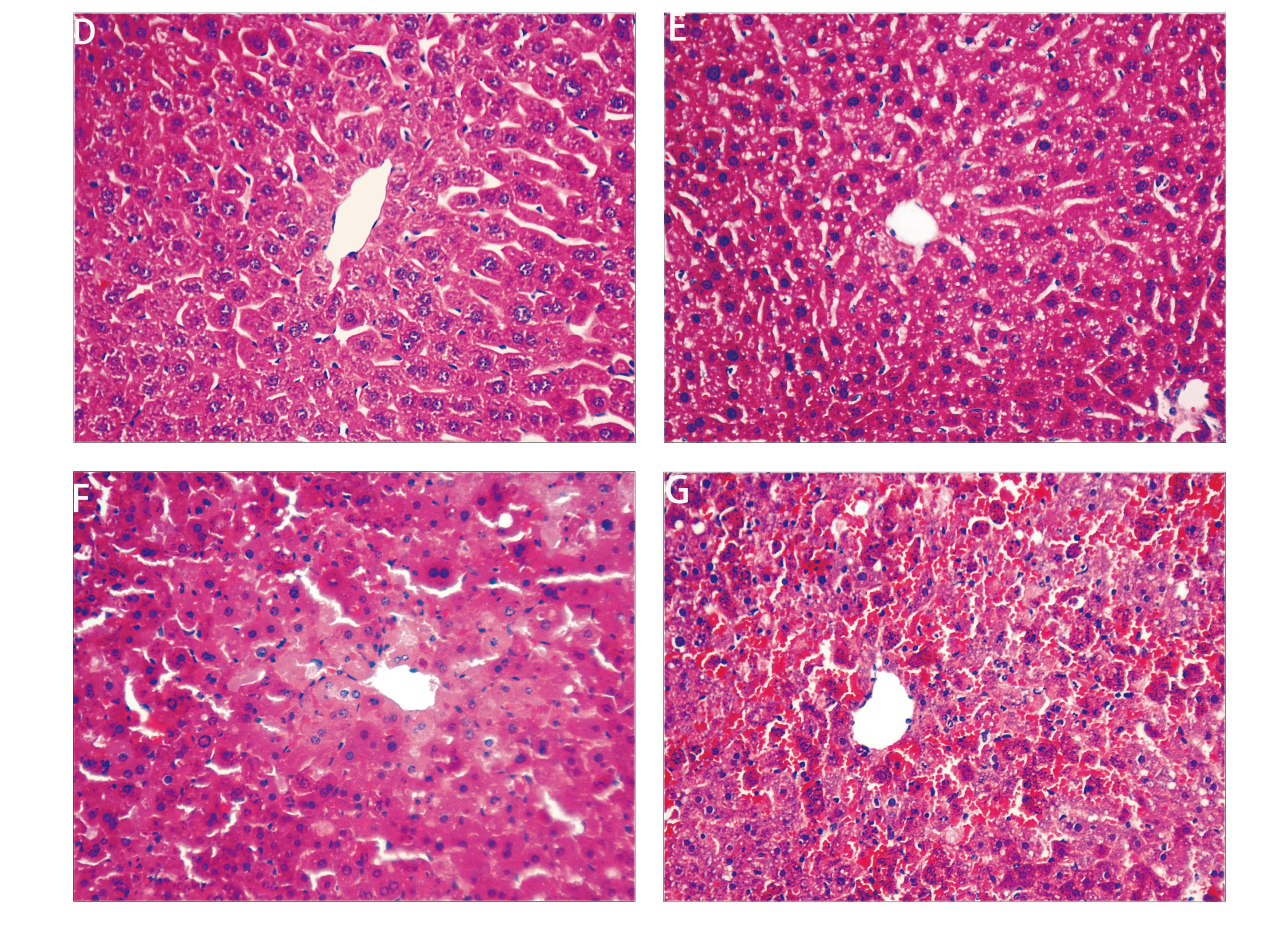
注:A为不同剂量组合的D-GalN/LPS注射后小鼠的24 h生存曲线;B为小鼠血清ALT和AST的动态变化;C为小鼠肝脏肉眼观的动态变化;D-G分别为0、3、5、7 h的小鼠肝组织(HE×400)
图1D-GalN/LPS诱导的急性肝衰竭建模成功
二、IL-33及其受体ST2在急性肝衰竭小鼠肝内的动态变化
如图2所示,随着D-GalN /LPS诱导后肝细胞损伤的加重,肝内IL-33 mRNA水平不断增高,在7 h肝细胞出现大块坏死并已达到肝衰竭时, 其表达水平达到高峰。而肝内ST2L受体蛋白含量在D-GalN /LPS诱导后3 h,肝细胞损伤的早期已经显著升高,之后开始不断下降,到7 h肝衰竭时下降至最低水平。
三、IL-33及其受体ST2在急性肝衰竭小鼠外周血清中的动态变化
如图3所示,外周血清中IL-33蛋白水平随时间持续升高,在7 h肝衰竭时达到最高水平,与IL-33 mRNA的动态变化相一致。与此不同的是,血清中ST2蛋白水平在0 h和3 h肝细胞损伤的早期无明显差异,但在5 h肝细胞损伤的中期却出现显著的升高,之后又明显下降。
四、急性肝衰竭小鼠IL-33的肝内细胞来源
免疫组化结果显示,健康小鼠肝内IL-33主要定位于血管内皮细胞和肝血窦细胞核内(图4a)。肝衰竭小鼠肝脏内IL-33的细胞来源也是这两种细胞,定位并未发生改变(图4b)。
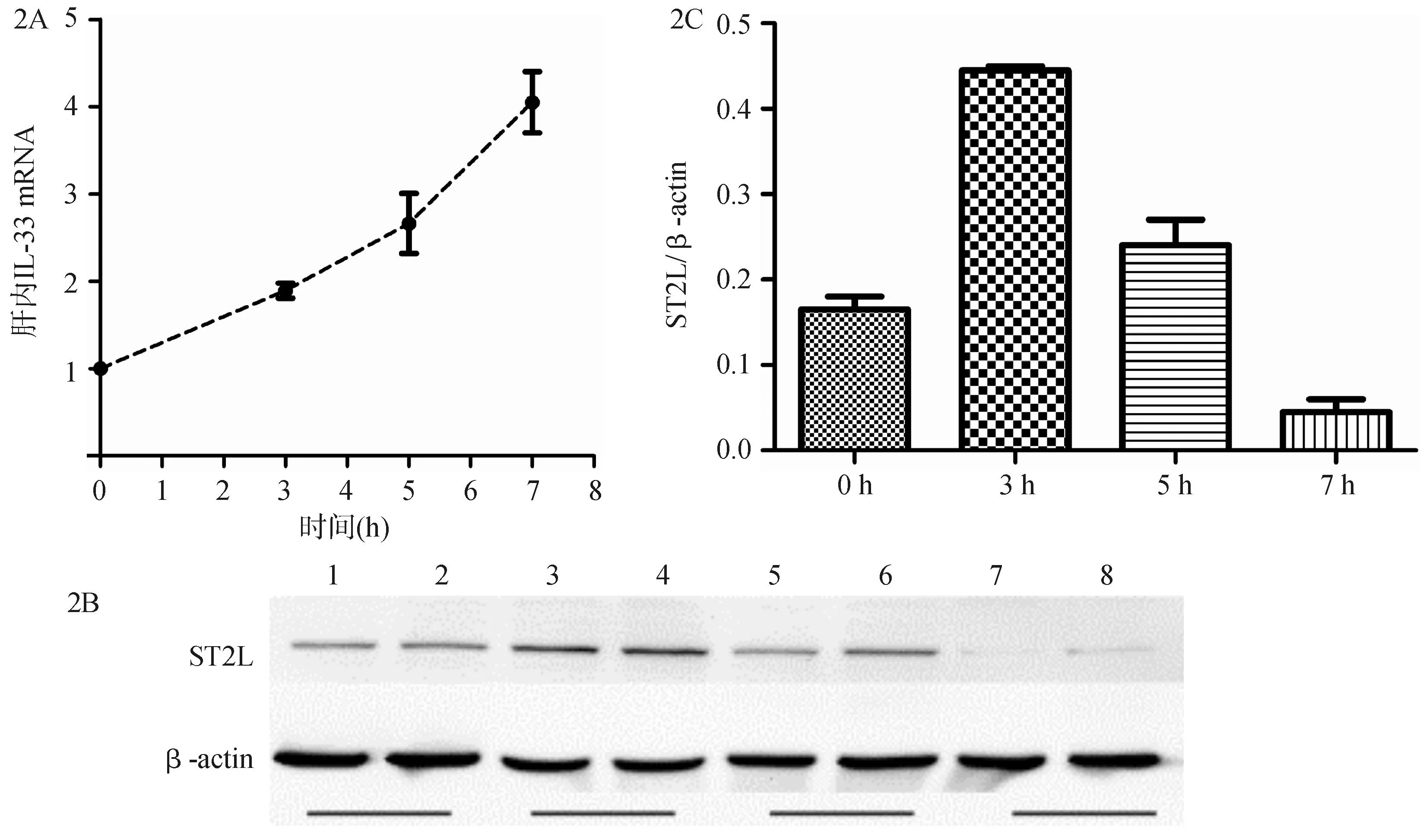
注:A.肝内IL-33 mRNA水平随时间逐步升高;B. 肝内ST2L蛋白含量在3 h增加明显,之后逐步减少,至7 h达最低水平;C. 肝内ST2L蛋白Western 印迹对应的灰度直方图
图2肝内IL-33和ST2L随病情进展的动态变化
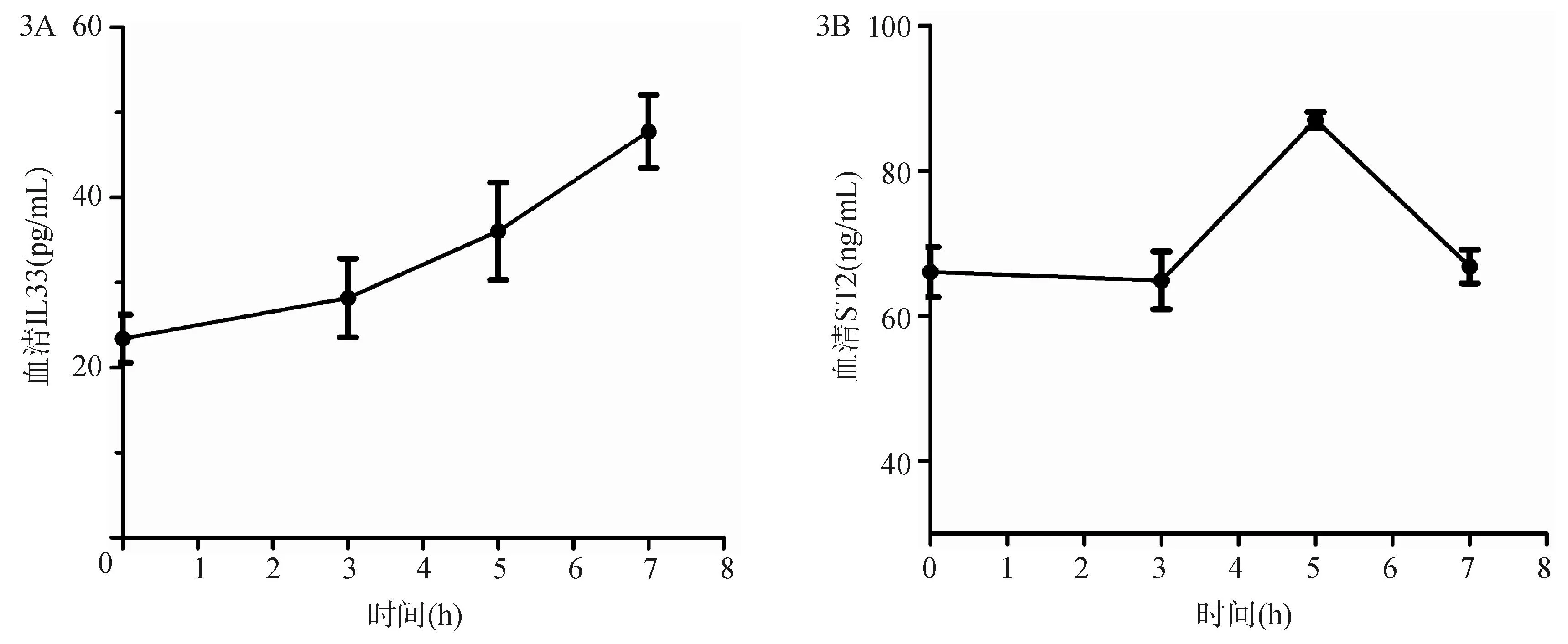
注:A. 血清IL-33水平逐渐攀升,至7 h达最高水平;B. 血清ST2在5 h出现峰值后转而下降
图3外周血清中IL-33和可溶型sST2蛋白随时间的动态表达谱
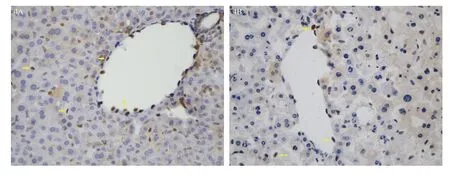
注:A. 健康小鼠肝内IL-33表达;B. 肝衰竭小鼠肝内IL-33表达
讨论
尽管近年来随着医疗技术的发展,急性肝衰竭患者的预后得到了一定改善,但目前病死率仍很高,其发生发展的具体机制亦不明确。全身炎性反应无疑在急性肝衰竭的发病过程中扮演至关重要的角色。IL-33是一种新近发现的、具有多种生物学效应的炎性细胞因子,属于IL-1超家族。研究证实,IL-33/ST2轴不仅可诱导Th2型免疫,参与过敏性疾病、纤维化疾病和寄生虫感染等疾病的发展过程[8, 10-12],还可通过促进Th1型免疫和CD8+T淋巴细胞在抗病毒和抗肿瘤免疫中发挥保护性作用[9, 13]。本研究通过D-GalN/LPS诱导的急性肝衰竭小鼠模型,发现IL-33及其受体ST2随时间动态变化与急性肝衰竭的病情进展存在紧密联系,说明IL-33/ST2轴参与了急性肝衰竭的发生发展过程,这或可为急性肝衰竭的治疗提供新的潜在靶点。
D-GalN/LPS被广泛用于急性肝衰竭发病机制以及治疗干预的研究。通过观察记录不同剂量组合的D-GalN /LPS腹腔注射后小鼠24 h生存曲线,发现D-GalN 900 mg/kg 联合LPS 10 μg/kg所对应的存活率与临床情况更为接近。
随着肝细胞损伤的不断加重,小鼠肝内IL-33 mRNA含量不断增高,至7 h肝组织表现为大块坏死时上升至峰值。而肝内ST2L蛋白在3 h肝细胞损伤的早期即达高峰,随后持续减少,在7 h肝衰竭时降至最低。外周血清中IL-33蛋白水平的动态变化与肝内IL-33 mRNA变化相吻合,也随时间不断升高,在7 h达高峰。血清中ST2蛋白水平在0 h和3 h肝细胞损伤的早期无明显差异,但在5 h肝细胞损伤的中期却出现明显升高,之后显著降低。研究证实,IL-33有两种存在形式:IL-33前体和IL-33成熟体,分别位于细胞核内和细胞外。IL-33的受体ST2也至少有两种亚型:跨模型ST2L与可溶型sST2,前者存在于靶细胞膜上,后者则存在于外周血清中。随着肝衰竭病情进展,肝细胞由点状、小片状的凋亡、坏死发展为大面积的凋亡、坏死,肝小叶结构崩塌,大量炎性细胞浸润,肝血窦充斥血细胞或闭塞不通,微循环障碍严重,因此IL-33前体作为一种警戒素被释放到细胞外。随时间推移,IL-33的血清水平逐步攀升。而表达于小鼠肝细胞膜上的ST2L随着肝细胞坏死释放入血[14],成为血清中sST2的一部分,所以在5 h肝内ST2L开始减少,而sST2水平在5 h出现一次跃升。之后在7 h,肝内ST2L进一步减少,可溶型sST2下降。这或许与重型肝炎时出现的“胆酶分离”原理类似,疾病末期肝细胞大量坏死,肝内合成ST2L的能力减弱,而血清中的sST2部分被降解,引起sST2水平的下降。
IL-33在肝脏内的细胞来源可因疾病状态的差异而出现不同。肝细胞肝癌时,IL-33主要定位于效应记忆性CD8+T淋巴细胞[9]。肝脏纤维化时,激活的肝星状细胞为IL-33的主要细胞来源[8]。而在Con-A诱导的小鼠急性肝损伤模型中,IL-33则定位于肝细胞核内[15]。健康小鼠肝内IL-33主要表达于血管内皮细胞和肝血窦细胞核内,肝衰竭肝脏内IL-33的细胞来源也是这两种细胞,定位并未发生改变。
在急性肝衰竭的发展过程中,TNF-α、IL-1β、IL-6和IL-18等引发炎性级联反应,造成肝细胞大量凋亡,最终导致多器官功能衰竭。在Con-A诱导的小鼠急性肝损伤中,注射IL-33可下调TNF-α、IFN-γ和IL-17等促炎因子的血清水平,减轻产生这些细胞因子的免疫细胞在肝内的浸润程度,包括CD4+T淋巴细胞、CD8+T淋巴细胞和NK细胞[16]。还可抑制促凋亡蛋白caspase-3和BAX的激活,同时上调抗凋亡蛋白Bcl-2和p-ERK的表达,从而缓解肝损伤。IL-33可通过激活NF-κB和p38 MAPK信号通路,增强Bcl-2和cyclin D1的蛋白表达,从而在肝脏缺血缺氧损伤中发挥重要的保护作用[14]。肝衰竭时肠道的屏障功能受到削弱,肠菌移位,引起内毒素血症,形成二次打击,进一步加重了肝衰竭。肝衰竭时高水平的促炎因子,如TNF-α和IL-6,可诱导sST2的生成[17]。一方面,过表达的血清sST2已被证明能与巨噬细胞相结合,通过下调Toll样受体4(TLR4),抑制促炎因子(IL-6、IL-12和TNF-α等)的表达和Th1型细胞反应,有助于对内毒素血症形成耐受[18-19]。另一方面,血清sST2作为一种可溶性诱饵受体,可中和外周游离的IL-33,防止IL-33全身性效应的发生[6]。动物实验显示,若通过基因改造使IL-33染色质锚定结构缺失,小鼠将形成高IL-33血症,引起多器官严重的炎性细胞浸润,最终导致致死性炎症[20]。
综上所述,IL-33及其受体ST2随时间的动态变化与急性肝衰竭的病情进展间存在紧密的联动关系,说明IL-33/ST2轴参与了急性肝衰竭的发生发展过程。IL-33/ST2轴有望成为急性肝衰竭临床治疗的潜在靶点,为救治急性肝衰竭患者提供希望。
参考文献
1 Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology, 2005, 41:1179-1197.
2 Bernal W, Auzinger G, Dhawan A, et al. Acute liver failure. Lancet, 2010, 376:190-201.
3 Guo LM, Liu JY, Xu DZ, et al. Application of molecular adsorbents recirculating system to remove NO and cytokines in severe liver failure patients with multiple organ dysfunction syndrome. Liver Int, 2003, 23 Suppl 3:16-20.
4 Roth GA, Faybik P, Hetz H, et al. Pro-inflammatory interleukin-18 and Caspase-1 serum levels in liver failure are unaffected by MARS treatment. Dig Liver Dis, 2009, 41:417-423.
5Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity, 2005, 23:479-490.
6 Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol, 2011, 7:321-329.
7 Liang Y, Jie Z, Hou L, et al. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J Immunol, 2013, 190:5666-5675.
8 Marvie P, Lisbonne M, L'Helgoualc'h A, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med, 2010, 14:1726-1739.
9 Brunner SM, Rubner C, Kesselring R, et al. Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology, 2015, 61:1957-1967.
10Prefontaine D, Lajoie-Kadoch S, Foley S, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol, 2009, 183:5094-5103.
11Tajima S, Bando M, Ohno S, et al. ST2 gene induced by type 2 helper T cell (Th2) and proinflammatory cytokine stimuli may modulate lung injury and fibrosis. Exp Lung Res, 2007, 33:81-97.
12Humphreys NE, Xu D, Hepworth MR, et al. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol, 2008, 180:2443-2449.
13Bonilla WV, Frohlich A, Senn K, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science, 2012, 335:984-989.
14Sakai N, Van Sweringen HL, Quillin RC, et al. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology, 2012, 56:1468-1478.
15Arshad MI, Piquet-Pellorce C, L'Helgoualc'h A, et al. TRAIL but not FasL and TNFalpha, regulates IL-33 expression in murine hepatocytes during acute hepatitis. Hepatology, 2012, 56:2353-2362.
16Volarevic V, Mitrovic M, Milovanovic M, et al. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J Hepatol, 2012, 56:26-33.
17Kumar S, Tzimas MN, Griswold DE, et al. Expression of ST2, an interleukin-1 receptor homologue, is induced by proinflammatory stimuli. Biochem Biophys Res Commun, 1997, 235:474-478.
18Sweet MJ, Leung BP, Kang D, et al. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J Immunol, 2001, 166:6633-6639.
19Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol, 2004, 5:373-379.
20Bessa J, Meyer CA, de Vera Mudry MC, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun, 2014, 55:33-41.
(本文编辑:钱燕)
The expression profiles and significance of IL-33 and its receptor ST2 in a murine model of D-GalN/LPS -induced acute liver failure
JIANGShao-wen,LINLan-yi,XIANGXiao-gang,LUJie,WANGFan,MORui-dong,LIUYu-han,CAIWei,WANGHui,XIEQing.DepartmentofInfectiousDisease,RuijinHospitalAffilicatedtoMedicalColledgofShanghaiJiaotongUniversity,Shanghai200025,China.
【Abstract】ObjectiveTo investigate the expression profiles and implication of IL-33 and its receptor ST2 in a murine model of acute liver failure (ALF) induced by D-GalN/LPS. MethodsThe ALF murine model was set up by intraperitoneal injection of D-GalN (900 mg/kg)/LPS(10 ug/kg), and confirmed by histopathology and biochemistry. Dynamic expression profiles of IL-33 and its receptor ST2 in ALF murine model were investigated by quantitative polymerase chain reaction (q-PCR), western-blot, enzyme-linked immune-sorbent assay (ELISA) and immunohistochemistry at different time points, respectively. ResultsThe murine model of ALF was successfully established by intraperitoneal injection of D-GalN (900 mg/kg)/LPS(10 ug/kg). The mRNA level of intra-hepatic IL-33 continuously increased with the progression of ALF, and reached the peak at 7 h after D-GalN/LPS challenge. Compared to baseline, intra-hepatic ST2L protein level was markedly up-regulated at 3 h, which was before the occurrence of obvious hepatocytes damage. However, it declined later on and fall to the lowest at 7 h. In addition, it was observed that IL-33 protein level in serum was elevated sustainedly over time and reached the highest level at 7 h, which was consistent with its dynamic mRNA changes. In contrast, the serum level of sST2 had no significant differences between 0 h and 3 h at the early stage of liver damage, but showed an obvious rise to climax at 5 h in the medium-term of liver damage, and then was followed by a drastic decline. The immunohistochemistry results confirmed that intra-hepatic IL-33 was mainly located in the nucleus of endothelial cells and sinusoidal cells in ALF mouse liver. ConclusionThe dynamic changes of IL-33 and its receptor ST2 in ALF mice are closely linked with the progression of acute liver failure, suggesting that IL-33/ST2 axis is indeed involved in the process of ALF. This might provide a novel potential target for ALF treatment.
【Key words】IL-33; ST2L; sST2; ALF; Dynamic expression
(收稿日期:2015-12-31)
Corresponding author:XIE-Qing,Email:xieqingrjh@163.com
通信作者:谢青,Email:xieqingrjh@163.com
基金项目:国家自然科学基金(81171569, 81300316, 81570535),国家十二五重大专项(2012ZX10002003-003, 2012ZX10002007-002, 2012ZX10002007-003-008),国家临床重点专科建设项目 (感染病学),上海市卫生和计划生育委员会课题(20144329),上海市学科带头人计划(12XD1403600),中国肝炎防治基金会王宝恩肝纤维化研究基金项目(CFHPC20131056)。
猜你喜欢
传染病信息(2022年6期)2023-01-12
中老年保健(2022年4期)2022-11-25
肝博士(2022年3期)2022-06-30
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年3期)2021-12-03
当代水产(2019年9期)2019-10-08
医学研究杂志(2015年2期)2015-06-10
