
煤灰中高岭石熔融性的密度泛函理论研究
2016-04-15 03:06:16许鲁霞杜梅芳李瑞连
上海理工大学学报 2016年1期
许鲁霞, 杜梅芳, 李瑞连, 江 健
(上海理工大学 理学院,上海 200093)
煤灰中高岭石熔融性的密度泛函理论研究
许鲁霞,杜梅芳,李瑞连,江健
(上海理工大学 理学院,上海200093)
摘要:选出两种复盐,熔融温度高的高岭石和熔融温度低的含钾高的微斜长石.以第一性原理的密度泛函理论(DFT)和量子化学从头算法为理论基础,结合PW 91算法和广义梯度近似(GGA),对煤灰中高岭石和微斜长石的反应活性进行计算,然后分析它们的最高能量占据轨道(HOMO)、最低空轨道(LUMO)、态密度和Milliken布局数.计算结果表明:高岭石轨道的能级差ΔE较大、化学活性低,高温脱水后生成莫来石,莫来石的结构比较稳定;高岭石脱水后易和电子接受体反应,不易和电子给予体反应,K+作为电子接受体进入高岭石晶格变成微斜长石,煤灰熔融温度降低.
关键词:量子化学; 高岭石; 反应活性; 微斜长石; 煤灰
准东煤是我国近几年发现的大型煤田,它的预测储量已经达到了将近4 000亿 t,是新疆及周边地区的主要煤源[1].但是,由于准东煤高水分、钠钾盐含量高的特殊性质,以至于在燃烧时炉内燃烧器区出现了严重的结渣粘污问题,其中,包含了复杂的物理化学变化[2].煤灰中主要矿物质熔融过程中的物理化学变化和相互作用机理对煤灰的熔融特性有着非常重要的影响.高温下矿物质熔融性与煤灰中的灰分构成有关,通常情况下,酸性氧化物具有提高煤灰熔融温度的作用,其含量越多,熔融温度就越高,如Al2O3,SiO2,TiO2等;相反,碱性氧化物与煤灰的熔融温度成反比,如Fe2O3,CaO,MgO,Na2O和K2O等[3].Vassilev等[4]分析了多种煤的结构及其对应煤灰的化学构成,并以此探讨了煤灰中主要物质组成和灰熔融性的关系后发现,灰熔融温度与长石、氯化物、蒙脱石和硫化物的含量成反比,它们的含量越高,熔融温度越低;灰熔融温度和金红石、莫来石和高岭石的含量成正比.
第一性原理方法也叫从头计算的方法,是一种理论性比较强的计算方法,是基于密度泛函理论的.伴随量子化学理论的发展以及量子化学计算方法和技术的进步,国内外学者已经可以运用半经验算法及从头计算的方法去探索物质进一步的物化性质、结构特点、作用机理及活性位点等,进而从微观的层面上解释物质的宏观物理特性[5].深入探索煤灰中物质的性质尤其是它们的微观性质,就要更重视在微观的角度上研究煤灰中矿物质的熔融特性和锅炉受热面结渣的机理.在这一方面,已有大量文献基于宏观方面进行了深入讨论[6-8],在灰的熔融与矿物演变规律方面也有了深入的研究[9].微观的角度上,也有很多学者基于第一性原理计算了不同矿物质和氧化物的弹性常数或不同矿物质在(110)切面吸附能的大小,对锅炉燃煤过程中的粘污结渣进行有效预测[10-11].在我国新疆地区,煤储量非常丰富,但是,结渣问题却非常严重.这是因为煤中熔点低的钠钾盐含量太高,温度升高到一定程度时钠钾盐受热升华粘附在烟气中的固体颗粒上,最后冷凝并形成了结渣初始层,造成的粘污问题严重影响锅炉效率.现已有通过计算物质的最高能量占据轨道(HOMO)、最低空轨道(LUMO)和键长、布局数等性质,然后依次来判断物质其反应活性位点和转化机理等方面的探究[12-13].本文通过对高岭石和微斜长石态密度、Milliken布局数、HOMO和LUMO的计算,研究了高岭石高温下反应的微观机理,为进一步解决结渣问题提供理论基础.
1实验与计算方法
1.1实验方法
实验选用神木大柳塔煤,按照国标(GB 219-91)规定做成灰锥,然后利用灰熔点仪测定其特征温度.添加剂为高岭土(含有的主要矿物质为高岭石),按照不同的添加率(添加剂占煤灰的质量分数)加入添加剂,将其粉碎至粒度小于0.2 mm后,得到的混合物用玛瑙研钵研磨(粒度小于0.1 mm),最后按标准制成815 ℃(±10 ℃)灰的样本.按照国标(GB 154-79)分析煤灰和高岭石的化学组成,按照国标(GB 219-74),用HR-1型灰熔点测定仪在弱还原气氛中测定特征熔融温度[14].高岭石混合煤灰的特征温度如表1所示.
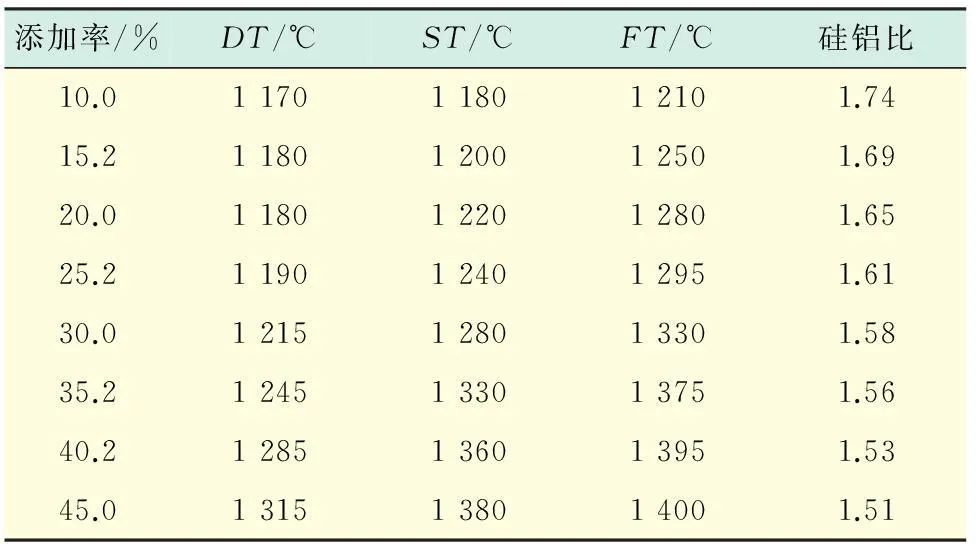
表1 高岭石混合煤灰的特征温度
由表1可知,随着高岭土添加率的增加,神木大柳塔煤灰的硅铝比从1.74降到1.51,而煤灰的特征温度DT(变形温度)、ST(软化温度)、FT(流动温度)却随之提高,当高岭石的添加率在30%左右时,神木大柳塔煤灰的ST和FT可提高至1 280 ℃和1 330 ℃.在这个熔融过程中,高岭石的反应为[15]
向煤中添加K2O时,会发生反应,出现熔融温度低的含钾复盐.
(3)
新疆准东煤(五彩湾煤)具有高水分、高挥发量、低灰分、低热值、强沾污及结渣严重的特点,和其它煤种的特性差异较大.燃烧时煤灰中K2O,Na2O含量严重偏高(其中,Na2O的含量高达2%,某些地域煤中含量甚至高达10%),在熔融过程容易和Al2O3,MgO,SiO2,FeO和Fe2O3形成低温共熔体或者生成钾、钠含量高的复盐,从而导致准东煤燃烧过程中锅炉在高温区域容易出现严重的粘污结渣现象.表2给出了一些共晶化合物的熔点[16].
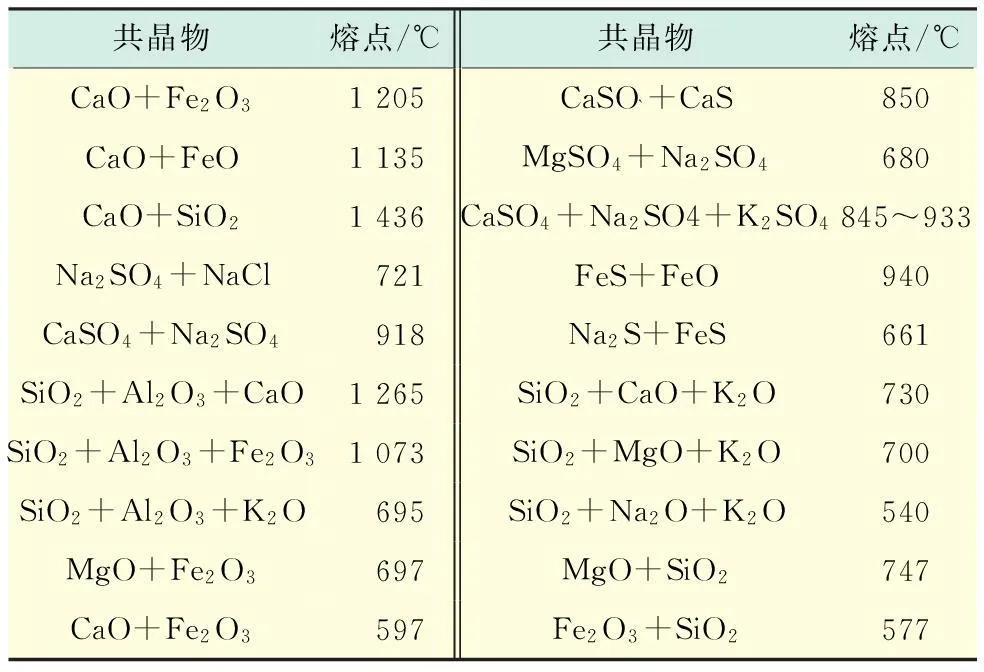
表2 共晶化合物熔点
1.2计算方法及理论
根据密度泛函理论(DFT)和量子化学从头算法,利用量子化学软件Material Studio里面的CASTEP模块构建高岭石和微斜长石的最稳定结构并计算它们的反应活性[17].第一性原理计算是通过粒子密度描述体系的基态性质,这一点与传统的量子理论利用波函数来描述是不同的.得出该理论的关键是要求解Kohn-Sham方程[18].
(4)
(5)
式中:hks为哈密顿量;r为所有电子坐标的集合;Rq为所有原子核坐标的集合;Zq为原子核电荷数;ni为占有数;φi(r)表示原子簇与分子的单电子波函数;ρ(r)为电荷密度.
在式(4)中第一项为电子的动能项;第二项为电子与原子核的库仑作用;第三项为Hartree势,可类比成库伦势;第四项Vxc是交互关联势.然后,在此基础上构建高岭石和微斜长石的最稳定结构,得出它们的结构参数.计算的交换相关能泛函采用了广义梯度近似(GGA)的PW 91(Wang和Peredew设计)[19-20]交换相关势.为了保证计算精度,总能量精度设置为1.0×10-5Ha,由于该体系考虑自旋与否对结果的影响不大,计算时忽略自旋极化,每个原子上的力小于0.02 Ha/nm,最大偏移小于0.000 5 nm,能量的收敛精度设为1.0×10-6.计算时,周期性边界条件选取高岭石和微斜长石晶体的一个原胞,采用Monkhost-Pack 方法,进行高对称性特殊点为(1×1×1)的布里渊区抽样,通过改变迭代次数使结构达到收敛.
2结构与讨论
2.1结构优化
高岭石(Al2O3·2SiO2·2H2O)属于三斜晶系,其空间群为P1(C1-1),经几何优化后的最稳定结构如图1(a)所示,晶格常数a=0.517 9 nm,b=0.898 7 nm,c=0.752 7 nm,晶面角α=91.242 5°,β=106.365°,γ=89.912 1°.高岭石的结构可以看成两部分,分别是4个硅氧四面体和4个AlO2(OH)4八面体.微斜长石属于三斜晶系,空间群为P1(C1-1),经几何优化后得到最稳定结构如图1(b)所示,晶格常数a=0.873 1 nm,b=1.329 4 nm,c=0.735 1 nm,晶面角a=90.535 1°,β=115.624°,γ=87.879 6°.微斜长石由12个AlO4和4个SiO4四面体构成,电荷平衡阳离子K+有4个位于晶面,另外2个镶嵌在四面体中.考虑到晶面、晶向和晶胞顶点对晶体单元的贡献分别为1/2,1/4,1/8,可以得到高岭石和微斜长石晶胞内有效原子个数.高岭石中Al,Si,O,H的原子个数分别为4,4,18,8,微斜长石中的Al,Si,O,K的原子个数分别为12,4,32,4.

图1 高岭石和微斜长石空间结构图
2.2高岭石的反应活性计算
计算几何优化后的高岭石的态密度、HOMO、LUMO和Milliken布局数,高岭石的(1×1×2)超晶胞的HOMO和LUMO如图2所示,总态密度(TDOS)如图3所示,Milliken布局数如表3所示.

图2 高岭石的HOMO和LUMO
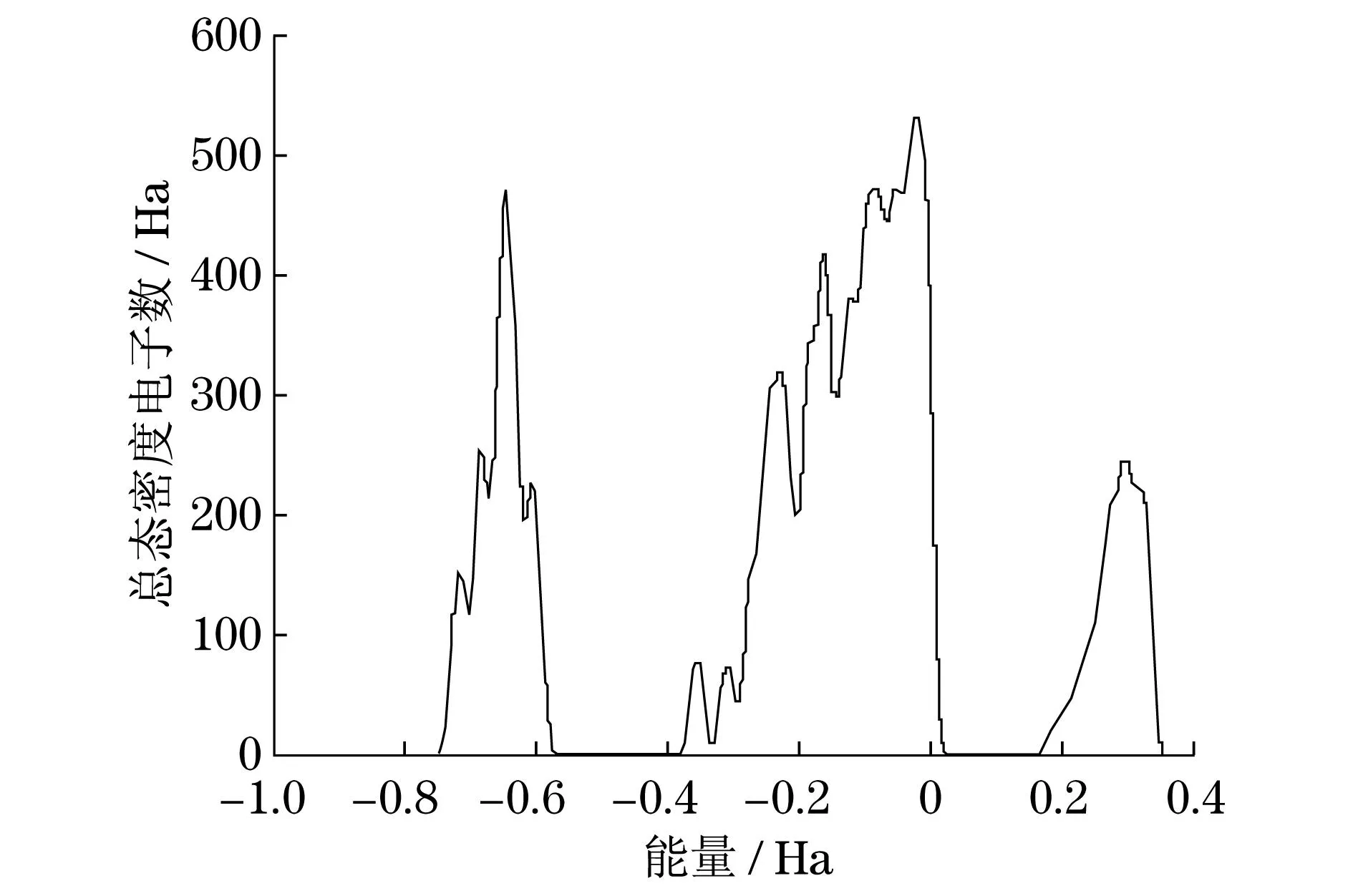
图3 高岭石的总态密度
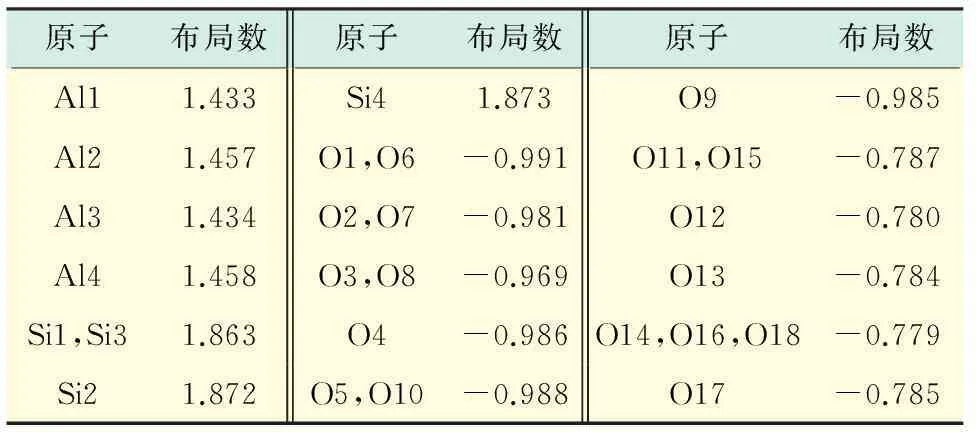
原子布局数原子布局数原子布局数Al11.433Si41.873O9-0.985Al21.457O1,O6-0.991O11,O15-0.787Al31.434O2,O7-0.981O12-0.780Al41.458O3,O8-0.969O13-0.784Si1,Si31.863O4-0.986O14,O16,O18-0.779Si21.872O5,O10-0.988O17-0.785
在前线分子轨道理论中,一般认为分子反应的实质是轨道的变化,其中,活性最高的是前线轨道.微观解释就是反应时外加电子会占据反应物的最低空分子轨道(LUMO),而物质若发生电离,电子会从反应物的最高被电子占有分子轨道(HOMO)转移,由此可以看出,可以根据前线轨道理论解释物质反应活性等性质.如图2所示,组成HOMO轨道的主要是Si-O四面体和OH里面的O,而且其轨道能级为-7.669 eV,具有较高的能态.当高岭石与其他物质反应时,Na+,K+等碱性氧化物中的亲电离子会与高岭石中组成HOMO的氧原子反应,使铝氧键断裂,电子发生位置变化而生成其他物质,因此,高岭石容易与电子接受体反应.从表3可以看出,O1,O2,O3,O4,O5,O6,O7,O8,O9,O10这些原子的净电荷分别为-0.991,-0.981,-0.969,-0.986,-0.988,-0.991,-0.981,-0.969,-0.985,-0.988,说明这些原子是HOMO的主要构成部分;O13,O14,O16,O17,O18这些原子所带的净电荷为-0.784,-0.779,-0.779,-0.785,-0.779,说明这些原子对HOMO的贡献较小.因此,当高岭石与电子接受体发生反应时,电子接受体容易从O1,O2,O3,O4,O5,O6,O7,O8,O9,O10原子所在的面进入晶格内.组成高岭石的LUMO的主要是Si和O,其轨道能级为-2.578 eV,数值较低,高岭石不易和亲核离子的体系反应.从表3可以看出,Si1,Si2,Si3,Si4,Al1,Al2,Al3,Al4原子所带的净电荷分别为1.863,1.872,1.863,1.873,1.433,1.457,1.434,1.458,这些原子对LUMO的贡献较大.比较电荷量发现,数值上Si原子的电荷量大于Al原子的电荷量,说明Si原子更容易与电子给予体反应.轨道的能级差ΔE为HOMO和LUMO能级的差值,其大小为-5.091 eV,其值较大,高岭石的反应活性较低.
图3为高岭石的总态密度(TDOS)图,费米能级(能量E=0)在左侧态密度范围内,且左侧峰值较高,右侧距离费米能级较远的为导带,且峰值较低,这表明了高岭石更倾向于和亲电体系(电子接受体)反应,不倾向于和亲核体系(电子给予体)反应,这与前面计算出的HOMO和LUMO的结果是一致的.
2.3微斜长石的反应活性计算
计算几何优化后的微斜长石的HOMO,LUMO,Milliken布局数.它的HOMO和LUMO如图4所示,部分键长数据如表4所示(见下页).
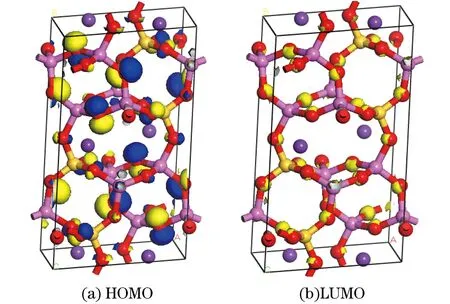
图4 微斜长石的HOMO和LUMO

键键长/nm键键长/nm键键长/nmO2-Al20.176101O2-Al30.175413O10-Al50.174715O10-Al30.175380O15-Al40.175413O15-Al50.176101O30-Al20.176101O21-Al80.176101O21-Al90.175413O26-Al80.174715O26-Al110.175380O30-Al10.175413O5-Si10.162756O6-Si10.162887O7-Si10.162516O12-Si20.162516O17-Si20.162756O19-Si30.162756O23-Si30.162516O28-Si40.162516O29-Si40.168870
如图4所示,组成微斜长石HOMO轨道的主要是O2,O8,O10,O15,O21,O25,O26,O30,其他氧原子对HOMO的贡献较小.HOMO的能级为-7.962 eV,能态较高,微斜长石容易和电子接受体作用成键.组成微斜长石LUMO轨道的同样是SiO4和AlO4中的氧原子,LUMO的能级为-2.576 eV,能态较低,微斜长石不易和电子给于体反应.由表4可以看出,O2,O8,O10,O15,O21,O25,O26,O30与Al原子成键时键长较大,当O5,O6,O7,O12,O17,O19,O23,O28,O29与Si原子成键时键长较小,这与HOMO计算结果是一致的.O2-Al2,O21-Al8,O15-Al5的键长最长,当微斜长石与电子接受体反应时,电子接受体进入微斜长石晶格内,使O2-Al2,O21-Al8,O15-Al5,O30-Al2键断裂,形成新的体系.
2.4高岭石的熔融机理分析
由表1可以看出,随着高岭石添加率的增加,煤灰的硅铝比降低,反映在CaO-Al2O3-SiO2相图中,煤灰组成逐渐移向莫来石区域,软化温度不断升高.当高岭石的添加率达到30%左右,神木大柳塔煤灰的软化温度和流动温度可提高至1 280 ℃和1 330 ℃.由前面的计算结果可知,高岭石ΔE偏大,不易反应.高温下高岭石中与H原子相连的O12,O13,O14,O16,O17,O18容易失去电子,高岭石脱H2O变为偏高岭石(Al2O3·2SiO2).在煤灰燃烧过程中,达到一定条件SiO2和Al2O3才重新组合形成莫来石(3Al2O3·2SiO2).之前对莫来石的反应活性已经有相关研究,莫来石的ΔE=0.224 76 Ha,其值较大,提供给分子达到活化态的能量需求比较大,更稳定,不易和其他物质反应.这从微观角度上给出了高岭石的添加降低了硅铝比并且使煤灰的熔融温度得到明显提高的原因.
由表2可以看出,KO2+Al2O3+SiO2,SiO2+Na2O+K2O,CaO+Fe2O3,Fe2O3+SiO2共晶物的熔点较低,分别为695,540,597,577 ℃.微斜长石的化学式为K2O·Al2O3·6SiO2,比较容易熔融.高岭石高温脱H2O后形成偏高岭石,K+从高岭石的O1,O2,O3,O4,O5,O6,O7,O8,O9,O10进入高岭石并与之结合,从而使其分子结构发生改变,最终生成微斜长石,降低了灰熔融温度.以上理论分析很好地验证了实验结果.
3结论
a. 以密度泛函理论为基础,通过第一性原理,计算高岭石和微斜长石的HOMO,LUMO,态密度和Milliken布局数等,分析高岭石的熔融特性.在高岭石左侧的态密度范围内,电子态密度的峰值最高,说明高岭石比较容易与电子接受体反应失去电子,相对于HOMO具有较大的能级;右侧导带距离费米能级较远且峰值低,说明高岭石不易和电子给予体反应,相对于LUMO具有较小的能级.
b. 高温下高岭石中与H原子相连的O12,O13,O14,O16,O17,O18容易失去电子,高岭石脱H2O变为偏高岭石(Al2O3·2SiO2),一定条件下SiO2和Al2O3重新组合生成莫来石(3Al2O3·2SiO2),提高了煤灰的熔融温度.当高岭石脱水后遇到亲电离子K+时,K+作为电子接受体从O1,O2,O3,O4,O5,O6,O7,O8,O9,O10原子所在的面进入高岭石晶格内,改变了高岭石的晶格结构,最终生成微斜长石,降低了灰熔融温度.
参考文献:
[1]杨忠灿,刘家利,何红光.新疆准东煤特性研究及其锅炉选型[J].热力发电,2010,39(8):38-40.
[2]陈川,张守玉,刘大海,等.新疆高钠煤中钠的赋存形态及其对燃烧过程的影响[J].燃料化学学报,2013,41(7):832-838.
[3]戴爱军.煤灰成分对灰熔融性影响研究[J].洁净煤技术,2007,13(5):23-26.
[4]Vassilev S V,Kitano K,Takada S,et al.Influence of mineral and chemical composition of coal ashes on their fusibility[J].Fuel Processing Technology,1995,45(1):27-51.
[5]Foresman J B,Frisch E.Exporing chemistry with electronic structure methods[M].2nd ed.Pittsburg,PA:Gaussian,Inc,1996.
[6]Zhang Y B,Tan Y W,Stormer H L,et al.Experimental observation of the quantum Hall effect and Berry’s phase in graphene[J].Nature,2005,438(7065):201-204.(编辑:丁红艺)
[7]Peres N M R.The electronic properties of graphene and its bilayer[J].Vacuum,2009,83(10):1248-1252.
[8]Sofo J O,Chaudhari A S,Barber G D.Graphane:a two-dimensional hydrocarbon[J].Physical Review B,2007,75:153401.
[9]乌晓江,张忠孝,周托,等.气化条件下混煤灰熔融特性及矿物质演变规律[J].燃烧科学与技术,2010,31(9):1590-1594.
[10]李明强,杜梅芳,乌晓江,等.锅炉结渣初始沉积层微观沉积机理研究[J].燃料化学学报,2013,41(2):157-162.
[11]杨换凌,张忠孝,乌晓江.高碱煤中NaCl与水冷壁吸附作用的量子化学研究[J].上海理工大学学报,2013,35(5):409-414.
[12]Hong H L,Min X M,Zhou Y.Orbital calculations of kaolinite surface:on substitution of Al3+for Si4+in the tetrahedralsites[J].Journal of Wuhan University of Technology-Mater,2007,22(4):661-666.
[13]李洁,杜梅芳,闫博,等.添加硼砂助熔剂煤灰熔融性的量子化学与实验研究[J].燃料化学学报,2008,36(5):519-523.
[14]王泉清,曾蒲君.高岭石对神木煤灰熔融性的影响[J].煤化工,1997(3):40-45.
[15]杨建国,刘志,赵虹,等.配煤煤灰内矿物质转变过程与熔融特性规律[J].中国电机工程学报,2008,28(14):61-66.
[16]李洁.用量子理论方法对煤灰微观结构特性的研究[D].上海:上海理工大学,2008.
[17]Vanderbilt D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Physical Review B,1990,41(11):7892-7895.
[18]朱维良,蒋华良,陈凯先,等.分子间相互作用的量子化学研究方法[J].化学进展,1999,11(3):247-253.
[19]Perdew J P,Wang Y.Accurate and simple analytic representation of the electron-gas correlation energy[J].Physical Review B,1992,45(23):13244-13249.
[20]Wang Y,Perdew J P.Spin scaling of the electron-gas correlation energy in the high-density limit[J].Physical Review B:Condensed Matter,1991,43(11):8911-8916.
(编辑:石瑛)
Density Functional Theory Regarding the Meltbility of Kaolinite in Coal Ash
XU Luxia,DU Meifang,LI Ruilian,JIANG Jian
(College of Science,University of Shanghai for Science and Technology,Shanghai 200093,China)
Abstract:Two kinds of double salts were selected:kaolinite with high melting temperature and microcline rich in potassium with lower melting temperature.The reaction activity of kaolinite and microcline was calculated based on the density functional theory (DFT) of First Principle and the quantum chemistry ab initio calculation by using the generalized gradient approximation (GGA) and the PW 91 algorithm.Then the hinghest occupied molecular orbital,lowest unoccupied molecular orbital,density of states and Milliken population of kaolinite and microcline were analyzed.The results show that the value of energy level difference ΔE of kaolinite is quite large which reduces the chemical reaction activity and mullite forms after kaolinite loses H2O molecular in high temperature.It is easy for kaolinite to react with the electron acceptor when it loses H2O molecular,while it is difficult for kaolinite to react with the electron donor.Kaolinite becomes microcline when K+as an electron accepter enters into the crystal lattice of kaolinite losing H2O molecular which lowers the melting temperature of coal ash.
Keywords:quantum chemistry; kaolinite; reaction activity; microcline; coal ash
中图分类号:O 641
文献标志码:A
通信作者:杜梅芳(1959-),女,副教授.研究方向:凝聚态物理.E-mail:dumeif@163.com
基金项目:国家自然科学基金资助项目(51276212)
收稿日期:2014-10-30
DOI:10.13255/j.cnki.jusst.2016.01.013
文章编号:1007-6735(2016)01-0076-05
第一作者: 许鲁霞(1991-),女,硕士研究生.研究方向:煤灰中矿物质的第一性原理计算研究.E-mail:xulux0802@163.com
猜你喜欢
煤质技术(2024年1期)2024-03-18 09:37:06
应用化工(2022年11期)2022-12-21 08:16:26
煤化工(2021年3期)2021-07-14 07:25:30
矿产综合利用(2020年1期)2020-07-24 08:51:26
山东冶金(2018年6期)2019-01-28 08:14:42
广西科技大学学报(2016年1期)2016-06-22 13:10:38
化工进展(2015年6期)2015-11-13 00:31:59
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:47
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:47
中国新技术新产品(2014年5期)2014-07-30 09:40:22
