
HGF/c-Met信号通路在克唑替尼诱导不同肺癌细胞株凋亡中的作用*
2016-04-15 09:05吕金益董芷辛李娅妮宁瑞玲宋向群周韶璋
中国病理生理杂志 2016年3期
吕金益, 董芷辛, 李娅妮 , 宁瑞玲, 宋向群, 周韶璋△
(广西医科大学 1附属肿瘤医院, 2研究生院,广西 南宁 530021)
HGF/c-Met信号通路在克唑替尼诱导不同肺癌细胞株凋亡中的作用*
吕金益1,董芷辛1,李娅妮1,宁瑞玲2,宋向群2,周韶璋2△
(广西医科大学1附属肿瘤医院,2研究生院,广西 南宁 530021)
[摘要]目的: 观察克唑替尼(crizotinib)诱导不同肺癌细胞株凋亡中HGF/c-Met信号通路的变化并探讨其调控机制。方法:采用噻唑蓝(MTT)法检测克唑替尼对H1993(c-Met扩增的肺腺癌细胞)、H2228(含有EML4-ALK融合基因的肺癌细胞)和A549细胞的活力抑制情况;采用流式细胞术检测3种细胞在克唑替尼作用后24 h、48 h和72 h的凋亡率;采用Western blot检测细胞在克唑替尼作用前后HGF/c-Met信号通路中MET蛋白及其磷酸化形式p-MET的水平,同时观察其下游通路关键蛋白AKT、ERK、p-AKT和p-ERK的变化情况。结果:MTT结果表明克唑替尼作用72 h后,H1993、H2228和A549细胞株的细胞活力抑制率均呈剂量依赖性升高。流式细胞术检测发现随着克唑替尼作用时间的延长,细胞凋亡率呈时间依赖性增加(P<0.05)。Western blot检测结果提示在H1993细胞株和H2228细胞株中,p-MET、p-AKT和p-ERK随着时间的延长蛋白水平呈现下降趋势。而在A549细胞株中p-AKT、p-ERK和p-MET在药物作用72 h后的变化趋势不明显。结论:初步证实HGF/c-Met信号通路与克唑替尼诱导肺癌细胞株H1993和H2228凋亡相关。
[关键词]HGF/c-Met信号通路; H1993细胞; H2228细胞; 克唑替尼; 细胞凋亡
近十年来,基于分子靶点的个体化治疗在非小细胞肺癌(non-small cell lung cancer,NSCLC)研究中取得了重大进展,尤其是以表皮生长因子受体(epidermal growth factor receptor,EGFR)和间变淋巴瘤激酶(anaplastic lymphoma kinase,ALK)为靶点药物的发现,对NSCLC个体化治疗的发展具有里程碑式的意义,分子靶向治疗也成为越来越重要的研究方向。
克唑替尼(crizotinib)为针对ALK/c-Met双靶点的酪氨酸激酶抑制剂,主要用于治疗存在EML4-ALK融合基因的晚期NSCLC患者,且已取得显著疗效[1]。c-Met是一类原癌基因,其蛋白产物是肝细胞生长因子(hepatocyte growth factor,HGF)的受体,并具有酪氨酸激酶的活性,在与受体进行特异结合后可激活一系列的跨膜信号通路[2],从而促使上皮细胞出现增生、迁移等[3]。阻断HGF/c-Met系统的配对表达或信号转导可作为抗肿瘤侵袭和转移的治疗策略之一,因为阻断作用不仅能够抑制肿瘤生长,还能抑制肿瘤转移[4]。HGF及其c-Met受体因在NSCLC中的发生、发展及EGFR-TKI耐药中均有非常重要的作用[5-6],而成为NSCLC靶向治疗领域研究的一个重要方向。我们的研究通过克唑替尼作用在MET扩增的肺癌细胞系H1993细胞和EML4-ALK阳性的肺癌细胞系H2228细胞,观察克唑替尼诱导的细胞凋亡情况和HGF/c-Met通路及其下游信号的变化。
材料和方法
1实验材料
1.1细胞人非小细胞肺癌细胞H2228和H1993购自ATCC;人非小细胞肺癌细胞A549由广西医科大学肿瘤医学院实验部提供。
1.2主要试剂与仪器克唑替尼粉末制剂购于CST;RPMI-1640 培养基、胎牛血清和胰酶替代物购自Gibco;MTT 购自Amresco;Annexin V-PE/7AAD 细胞凋亡检测试剂盒购自BD;MET I 抗、p-MET I 抗、AKT I 抗、p-AKT I 抗、ERK I 抗和p-ERK I 抗均购自CST;BCA 试剂盒购自Merck。Western blot 仪器设备购于Bio-Rad。
2方法
2.1细胞培养按肿瘤贴壁细胞的常规培养方法培养H1993细胞、H2228细胞与A549细胞至良好状态。
2.2MTT法检测细胞活力按培养H1993细胞、H2228细胞和A549细胞至对数生长期,按(2~6)×103cells/well的密度接种于96孔板,预设置6个浓度,每个浓度设置4个复孔,置于细胞培养箱中培养24 h后,按浓度梯度加入药物,把细胞置于培养箱中继续培养72 h后,吸净孔内液体,加入MTT继续培养4 h后再加入二甲基亚砜(dimethyl sulfoxide,DMSO),充分振荡10 min,在492 nm波长下测量吸光度,分别计算出克唑替尼对H1993细胞、H2228细胞和A549细胞的IC50,实验重复3次。
2.3流式细胞术检测细胞凋亡和周期分布情况取生长状态良好的对数生长期H1993细胞、H2228细胞和A549细胞,以每孔4×105、6×105、8×105个细胞数量分别接种于6孔板中,培养24 h,其中H1993细胞加入克唑替尼浓度为200 nmol/L,H2228细胞加入300 nmol/L血清培养液,将按每孔4×105个接种的细胞培养72 h、按每孔6×105个接种的细胞培养48 h、按每孔8×105个接种的细胞培养24 h,以细胞凋亡试剂盒说明书收集细胞并染色后用流式细胞仪测定细胞凋亡率,实验重复3次。
2.4Western blot检测克唑替尼对MET/AKT/ERK信号通路相关信号蛋白表达的影响收集长满瓶的H1993细胞、H2228细胞和A549细胞,用RIPA裂解液提取细胞总蛋白,所得总蛋白经10%聚丙烯酰胺凝胶电泳转移至聚偏二氟乙烯(polyvinylidene difluoride,PVDF)膜上。MET I 抗稀释度1∶1 000,p-MET I 抗稀释度1∶1 000,AKT I 抗稀释度1∶2 000,p-AKT I 抗稀释度1∶1 500,ERK I 抗稀释度1∶1 000,p-ERK I 抗稀释度 1∶1 500,兔、鼠 II 抗稀释度1∶2 000,以β-肌动蛋白(β-actin)为内参照。ECL发光底物显色,扫描分析条带,以条带灰度确定蛋白表达,实验重复3次。
3统计学处理
实验结果采用SPSS 16.0统计软件进行分析。各组计量资料数据用均数±标准差(mean±SD)表示,多组数据比较采用单因素方差分析,各组均数间的两两比较采用SNK-q检验和LSD法。以P<0.05为差异有统计学意义。
结果
1MTT比色法检测克唑替尼对H1993细胞、H2228细胞和A549细胞株生长的影响
MTT实验计算得出克唑替尼对H1993细胞的IC50为179 nmol/L,对H2228细胞的IC50为335 nmol/L。A549细胞对克唑替尼的处理不敏感,给予10倍H2228细胞的药物剂量未能求出IC50。H1993细胞、H2228细胞和A549细胞的增殖抑制率都随着克唑替尼药物浓度的升高相应增加,且呈剂量依赖性,见图1。
2流式细胞术检测细胞凋亡及周期分布
根据MTT所得IC50作为药物浓度,3组细胞未经过处理时凋亡率无显著差异,H1993细胞经克唑替尼作用24 h、48 h和72 h的凋亡率分别为(15.3±2.1)%、(27.2±1.6)%和46.5±1.8)%;H2228细胞经克唑替尼作用24 h、48 h和72 h的凋亡率为(13.7±0.8)%、(25.3±1.6)%和43.5±3.2)%;A549细胞经克唑替尼作用72 h的凋亡率为(15.64±0.61)%。药物作用后的H1993细胞和H2228细胞凋亡率较A549细胞和未加药处理组明显增加(P<0.05),且随时间延长凋亡率增加,表明H1993细胞和H2228细胞相对于A549细胞来说对克唑替尼更为敏感,见图2。
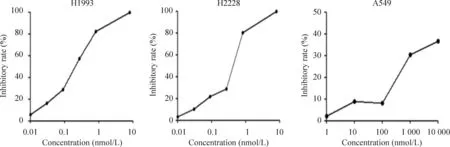
Figure 1.Crizotinib inhibited the viability of 3 cell lines of NSCLC. H1993, H2228 and A549 cells were treated with crizotinib at different concentrations (200 nmol/L and 300 nmol/L) for 72 h. The inhibitory rates were determined by the MTT assay. Mean±SD.n=3.
图1不同浓度的克唑替尼分别处理3种细胞72 h后的细胞活力抑制率
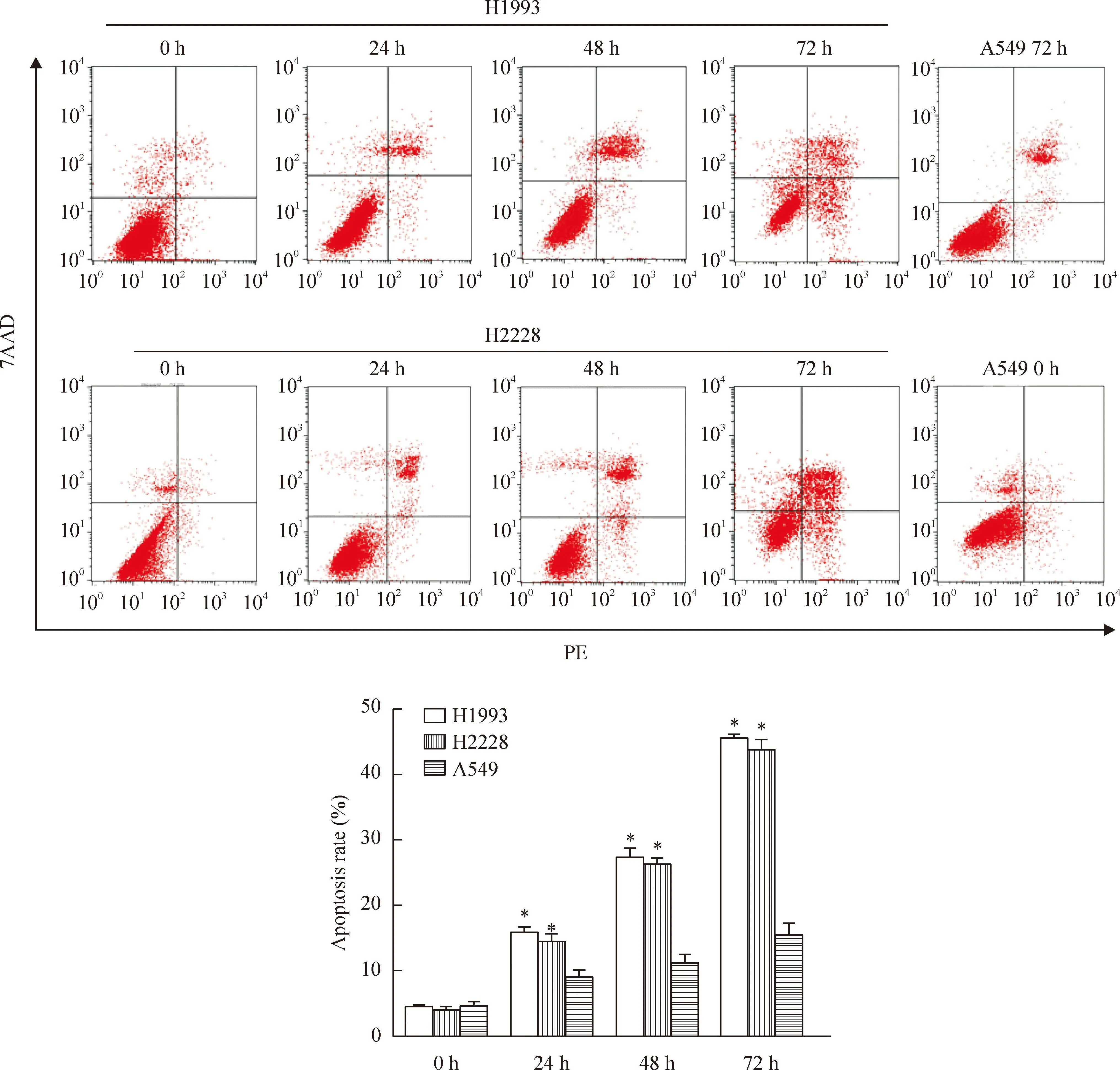
Figure 2.The apoptosis rates of the H1993, H2228 and A549 cells after treated with crizotinib at different time points. Mean±SD.n=3.*P<0.05vsA549.
图2经克唑替尼处理H1993、H2228和A549细胞后不同时点的细胞凋亡率
3Western blot检测克唑替尼对MET信号通路相关信号蛋白表达的影响
检测克唑替尼诱导H1993、H2228和A549细胞株凋亡中的蛋白变化。200 nmol/L克唑替尼处理c-Met扩增的H1993细胞,300 nmol/L 克唑替尼处理EML4-ALK阳性肺腺癌细胞株H2228细胞。在H1993细胞株中我们发现,经克唑替尼处理72 h后,MET总蛋白表达较未处理组降低,且其下游信号分子p-AKT、p-ERK和p-MET的蛋白水平在药物作用24 h后明显降低,并随时间延长不断下降,在72 h处p-MET的蛋白水平在三者之中最低。在H2228细胞株中,MET、AKT和ERK总蛋白表达无明显变化,但其下游信号分子p-AKT、p-ERK和p-MET的蛋白水平在药物作用48 h后有明显下降的趋势,在72 h时水平最低。而在相对不敏感肺癌细胞株A549中MET总蛋白及其下游信号分子p-AKT、p-ERK和p-MET在药物作用72 h后的蛋白水平变化趋势不明显,见图3。

Figure 3.The protein levels in the 3 cell lines treated with crizotinib at different time points (24 h, 48 h and 72 h) determined by Western blot. Mean±SD.n=3.*P<0.05vscontrol group (0 h).
图3经克唑替尼处理后的H1993、H2228和A549细胞在不同时点的蛋白检测结果
讨论
c-Met为原癌基因,是MET蛋白的编码基因。MET蛋白的配体为肝细胞生长因子(hepatocyte growth factor,HGF),正常的HGF/c-Met通路能调节胚胎发育及组织损伤修复,而异常的信号激活可促进细胞增殖、减少凋亡,同时使血管生成增多,肿瘤侵袭和转移行为增加[7]。c-Met与HGF结合引起c-Met胞质内酪氨酸残基的自身磷酸化,从而进一步激活下游几个重要的通路:PI3K-AKT信号通路、RAS-MAPK信号通路和STAT3通路[8]。在肺癌中,c-Met往往呈现出高表达的状态,而且提示与肿瘤的恶性程度有关[9]。克唑替尼为针对ALK/c-Met双靶点的酪氨酸激酶抑制剂,其作用于c-Met的机制主要是通过抑制c-Met激酶与ATP结合及两者结合之后的自身磷酸化而发挥作用。Zou等[10]通过细胞实验发现MET抑制剂克唑替尼能诱导细胞凋亡、减少细胞增殖及抑制血管生成; Okamoto等[11]应用克唑替尼对c-Met阳性与阴性的胃癌细胞进行研究,结果显示在c-Met阳性的胃癌细胞中c-Met信号通路AKT蛋白一同受到抑制进而诱导胃癌细胞的凋亡,而在c-Met阴性的胃癌细胞中却未观察到这一现象。郑时玉等[12]在乳头状甲状腺癌细胞实验中采用RNA干扰技术使c-Met沉默后发现肿瘤细胞的克隆形成、周期、迁移、侵袭能力都受到抑制。本研究中亦发现在使用克唑替尼作用c-Met扩增的H1993细胞和EML4-ALK阳性细胞株H2228时,其细胞抑制百分率呈浓度依赖性,细胞凋亡率随时间的延长而增加,两者相对于A549细胞和不加药对照组的凋亡率差异明显。这与以上学者研究结果相一致。在本研究中我们还发现c-Met水平的下降,可以推测克唑替尼是通过抑制MET蛋白来诱导细胞凋亡。另外,Ou等[13]报道1例存在c-Met扩增而非ALK融合基因的NSCLC患者使用克唑替尼后获得快速持续的缓解,提示克唑替尼在临床上可能作为一种c-Met抑制剂,但仍需要进一步研究。
AKT是PI3K/AKT/mTOR信号通路的关键分子,能激活下游底物mTOR及其下游p7056K、4E-BP1等信号因子,还能够通过磷酸化Bcl-2、Fox家族蛋白等来抑制细胞凋亡。ERK通路是目前研究较为深入的MAPK通路,ERK被酪氨酸激酶激活成p-ERK后进入细胞核,促进转录因子NF-κB、c-Myc等的磷酸化,促进细胞增殖及对药物诱导后的抗凋亡作用[14-15]。p-AKT及p-ERK是HGF/c-Met信号通路活化的主要标志,因此在本研究中我们采用Wes-tern blot的方法对MET蛋白和下游的AKT、ERK及其活化形式的信号蛋白进行检测,实验结果显示H1993细胞和H2228细胞在克唑替尼的作用下,MET、AKT和ERK蛋白的活化形式p-MET和p-AKT和p-ERK的蛋白水平随着时间的推移,均有不同程度下调,在72 h达到最低点。H1993细胞发生下调的时点早于H2228的下调时点,这可能与H1993细胞的c-Met扩增特性相关。而阴性对照A549细胞的MET、AKT和ERK总蛋白及其活化的p-MET、p-AKT和p-ERK蛋白均无明显受抑制作用,推测在本研究中,克唑替尼能通过抑制c-Met的活化形式p-MET来下调p-AKT和p-ERK磷酸化水平,从而抑制AKT和ERK通路促进细胞存活、抵抗凋亡的作用,促进肿瘤细胞的凋亡。这个研究结果也与Tanizaki等[16]和Kogita等[17]的实验结果相一致。
与此同时,在实验过程中我们也观察到,在药物作用72 h后,p-Met的表达基本被完全抑制,而其下游的信号蛋白p-AKT和p-ERK并未完全被抑制,我们推测在HGF/c-Met信号通路中可能还有其它的通路参与AKT、ERK信号通路的激活。已经有研究证实[18]c-Met可以通过p53信号通路抑制肺癌细胞的凋亡。Belalcazar等[19]研究发现c-Met信号通路与EGFR和HER3部分下游信号通路相通,存在“cross-talk”现象。Breindel等[20]也发现EGFR信号可以诱导MET的磷酸化, EGFR-MET的“cross-talk”现象不是直接发生的,而是由MET水平和中介信号通过ERK联合发生。在EGFR野生型或突变的非小细胞肺癌细胞中,抑制EGFR或ERK的信号通路可以减少MET的活化和蛋白水平表达。不过,具体是哪些信号通路或其它转录因子参与其中还有待更进一步的实验来证实。综上所述,我们可以认为HGF/c-Met通路在调控肿瘤细胞的生长方面有重要作用。
我们的研究为克唑替尼作为MET靶点的抑制剂在临床上的应用提供了证据。但由于克唑替尼诱导细胞凋亡机制复杂,不清楚的地方仍有很多。进一步研究HGF/c-Met信号的下游通路BIM和survivin,或对HGF/c-Met信号通路关键基因进行敲除,检测HGF/c-Met信号通路上各关键蛋白的表达水平和活化水平,明确通路上各蛋白之间的关系,可望更好阐明HGF/c-Met信号通路在肺癌细胞系凋亡中所起的作用。
[参考文献]
[1]Gandhi L, Janne PA. Crizotinib forALK-rearranged non-small cell lung cancer: a new targeted therapy for a new target[J]. Clin Cancer Res, 2012, 18(14):3737-3742.
[2]Yang M, Shan B, Li Q, et al. Overcoming erlotinib resistance with tailored treatment regimen in patient-derived xenografts from naïve Asian NSCLC patients[J]. Int J Cancer, 2013, 132(2):E74-E84.
[3]Sattler M, Salgia R. c-Met and hepatocyte growth factor: potential as novel targets in cancer therapy[J]. Curr Oncol Rep, 2007, 9(2):102-108.
[4]Stella MC, Trusolino L, Pennacchietti S, et al. Negative feedback regulation of Met-dependent invasive growth by Notch[J]. Mol Cell Biol, 2005, 25(10):3982-3996.
[5]Koudelakova V, Kneblova M, Trojanec R, et al. Non-small cell lung cancer: genetic predictors[J]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub, 2013, 157(2):125-136.
[6]Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC[J]. Cancer Cell, 2010, 17(1):77-88.
[7]Klapper LN, Kirschbaum MH, Sela M, et al. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors[J]. Adv Cancer Res, 2000, 77:25-79.
[8]Gelsomino F, Facchinetti F, Haspinger ER, et al. Targeting the MET gene for the treatment of non-small-cell lung cancer[J]. Crit Rev Oncol Hematol, 2014, 89(2):284-299.
[9]Park S, Choi YL, Sung CO, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients[J]. Histol Histopathol, 2012, 27(2):197-207.
[10]Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms[J]. Cancer Res, 2007, 67(9):4408-4417.
[11]Okamoto W, Okamoto I, Arao T, et al. Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive forMETamplification[J]. Mol Cancer Ther, 2012, 11(7):1557-1564.
[12]郑时玉,王丽,刘泽兵,等. 慢病毒介导的c-met RNA干扰对人乳头状甲状腺癌K1细胞生物学行为的影响[J]. 中国病理生理杂志, 2012,28(10):1819-1824.
[13]Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novoMETamplification[J]. J Thorac Oncol, 2011, 6(5):942-946.
[14]Janmaat ML, Giaccone G. The epidermal growth factor receptor pathway and its inhibition as anticancer therapy[J]. Drugs Today (Barc),2003, 39(Suppl C):61-80.
[15]Zimmer S, Kahl P, Buhl TM, et al. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling[J]. J Cancer Res Clin Oncol, 2009, 135(5):723-730.
[16]Tanizaki J, Okamoto I, Okamoto K, et al. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer accor-ding toMETalterations[J]. J Thorac Oncol, 2011, 6(10):1624-1631.
[17]Kogita A, Togashi Y, Hayashi H, et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK-positive non-small cell lung cancer[J]. Int J Oncol, 2015, 46(3):1025-1030.
[18]Liu Y, Liu JH, Chai K, et al. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells[J]. J Pharm Pharmacol, 2013, 65(11):1622-1642.
[19]Belalcazar A, Azana D, Perez CA, et al. Targeting the Met pathway in lung cancer[J]. Expert Rev Anticancer Ther, 2012, 12(4):519-528.
[20]Breindel JL, Haskins JW, Cowell EP, et al. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis[J]. Cancer Res,2013,73(16):5053-5065.
(责任编辑: 陈妙玲, 罗森)
阻断BMK1通路可通过调控BNIP3和BNIP3L抑制肿瘤干细胞
肿瘤干细胞(CSCs)具有干细胞相关的许多特性,并被认为可操控肿瘤的发生。尽管靶向CSCs具有开发新药物的巨大潜力,但由于目前缺乏有效的药物靶点和合适的药理学制剂,其发展仍有很大障碍。Song等的研究结果表明,BMK1的磷酸化不仅与胚胎干细胞和诱导性多能干细胞有关,也与CSCs有关。通过表达MEK5D激活BMK1,可增强CSCs的自我更新(微球体形成实验证实之)、增殖(克隆形成实验证实之)和成瘤能力,而BMK1抑制剂XMD8-92能抑制上述过程。RNA测序和微阵列分析结果表明,抑制BMK1显著增强了细胞死亡过程中的重要蛋白BNIP3和BNIP3L的表达。用shRNA沉默BNIP3和BNIP3L削弱了BMK1抑制剂XMD-8-92对CSCs微球体形成和克隆形成能力的抑制作用。以上结果说明BMK1对维持CSCs的“干性”起到关键性作用,提示BMK1有可能成为针对CSCs的药物靶标。
Oncotarget, 2015, 6(32):33279-33289(黄雪)
Role of HGF/c-Met signaling pathway in crizotinib-induced apoptosis of different lung carcinoma cell lines
LÜ Jin-yi1, DONG Zhi-xin1, LI Ya-ni1, NING Rui-ling2, SONG Xiang-qun2, ZHOU Shao-zhang2
(1AffiliatedTumorHospital,2PostgraduateCollege,GuangxiMedicalUniversity,Nanning530021,China.E-mail:zhoushaozhang@qq.com)
[ABSTRACT]AIM: To investigate the role of HGF/c-Met signaling pathway in crizotinib-induced apoptosis of different lung carcinoma cell lines and to analyze its potential regulatory mechanisms. METHODS: EML4-ALK positive cell line H2228, c-Met proliferation cell line H1993 and control cell line A549 were treated with crizotinib at different doses for different time periods. The viability of the cell lines was measured by MTT assay. The apoptosis was analyzed by flow cytometry with PI staining. The protein levels of MET and phosphorylated MET (p-MET) of HGF/c-Met signaling pathway as well as its down-stream key proteins AKT, ERK, p-AKT and p-ERK in the cell lines before and after crizotinib treatment were examined by Western blot. RESULTS: The growth of H1993, H2228 and A549 cell lines was inhibited after crizoti-nib treatment for 72 h in a dose-dependent manner. Apoptotic rates of H1993 cells and H2228 cells were increased with the crizotinib concentration and exposure time. Down-regulation of p-MET, p-AKT and p-ERK at protein levels in H1993 cells and H2228 cells after exposure to crizotinib for 72 h was confirmed by Western blot. No obvious change of the related-proteins of HGF/c-Met signaling pathway was found in A549 cell line. CONCLUSION: HGF/c-Met signaling pathway may contribute to crizotinib-induced apoptosis of H1993 cells and H2228 cells, which provides the experimental basis for MET-targeting treatment of lung cancer.
[KEY WORDS]HGF/c-Met signaling pathway; H1993 cells; H2228 cells; Crizotinib; Apoptosis
doi:10.3969/j.issn.1000- 4718.2016.03.010
[中图分类号]R730.23
[文献标志码]A
通讯作者△Tel: 0771-5334955; E-mail: zhoushaozhang@qq.com
*[基金项目]国家自然科学基金资助项目(No.81260357; No.81060188)
[收稿日期]2015- 11- 04[修回日期] 2015- 12- 23
[文章编号]1000- 4718(2016)03- 0445- 06
杂志网址: http://www.cjpp.net
猜你喜欢
中国药房(2022年12期)2022-07-02
锦州医科大学报(2022年1期)2022-04-20
医学信息(2019年21期)2019-12-05
人人健康(2019年3期)2019-04-02
中国现代医学杂志(2018年23期)2018-08-20
中国中药杂志(2016年22期)2017-02-13
科技视界(2016年15期)2016-06-30
中国实用医药(2016年11期)2016-05-04
山东体育学院学报(2015年3期)2015-08-14
中国当代医药(2015年18期)2015-08-06
