
家族性神经纤维瘤病Ⅰ型1例*
2016-04-13 09:44张波,吴国敏,徐澍
贵州医科大学学报 2016年1期
关键词:免疫组织化学
·病例报道·
家族性神经纤维瘤病Ⅰ型1例*
张波1, 吴国敏2, 徐澍3**
(1.德江县民族中医院 病理科, 贵州 德江565200; 2.贵州医科大学附属口腔医院 病理科, 贵州 贵阳550004; 3.贵州医科大学附院 病理科, 贵州 贵阳550004)
[关键词]神经系统疾病; 神经纤维瘤病; 免疫组织化学
神经纤维瘤病Ⅰ型(neurofibromatosis type 1,NFⅠ)又名冯.雷克林豪森病(von Recklinghausen’s disease),由von Recklinghausen于1882年首次描述。NFⅠ型为常染色体显性遗传病,外显率极高, 基因定位于染色体17qll.2,患病率约为(30~40)/10万。NFⅠ型确切的发病机制不是很清楚,认为除与NF1基因突变相关之外,可能还与激素、端粒酶、促血管生成因子、肿瘤微环境和电生理改变等多种肿瘤促发因素有关。NF1基因表达丢失,导致神经纤维蛋白Ras GAP功能丧失,最终导致Ras活性增加、细胞增生以及肿瘤形成,使受累个体发生良性及恶性肿瘤的机率增加[1]。国内文献报道的家族性NFI型患者较少,现将2015年4月收治的1例家族性NFI型患者临床资料报道如下。
1临床资料
1.1一般资料
患者女性,17岁。因发现左上睑凸起并逐渐增大17+月,于2015年04月入院。患者约出生3个月时家属无意间发现左眼有一“针尖”大小凸起,未予重视;逐渐增大,无疼痛、破溃、流脓,肿块逐渐侵犯眼球致左眼不能睁、完全失明,左眼上睑板外翻;曾就诊多家医院,诊断不明,未予任何治疗,于2013年就诊于外院行MRI检查示:(1)左侧睑面部及左眼眶内病变,考虑先天性病变、血管瘤或血管淋巴管瘤;(2)左侧晶状体未见确切显示,请结合临床;(3)左侧上颌窦炎。未行治疗。本次就诊后以“神经纤维瘤?血管瘤?血管淋巴管瘤?”收入院。家族中患者外婆、姨妈、母亲及姨表姐妹均有全身大小不等的多发性牛奶咖啡斑,但无其他相关改变。否认家族中男性患此病。体查:见左侧颜面部12 cm×10 cm×3 cm肿物,表面皮肤正常,与周围组织界限欠清,质中,稍可推动,无触痛;左眼不能睁、完全失明,左眼上睑板外露,右眼无异常;近发际线处延伸至头皮约12 cm×5 cm皮肤色黑、点状凸起,似“牛奶咖啡斑”,在患者腰及四肢可见大小不等的“牛奶咖啡斑”,表面毛发生长正常。彩色多普勒超声检查示左侧眼睑部皮下探及低回声团33 mm×19 mm×28 mm,边界尚清楚,形态不规则,团块内回声不均质,可见多发结节。CDFI示团块内可见条状血流信号,PW呈低速低阻动脉频谱。彩超诊断示左侧眼睑部皮下低回声团,具良性征象。MRI检查示:(1)左侧睑面部及左眼眶内病变,考虑先天性病变,血管瘤或血管淋巴管瘤;(2)左侧晶状体未见确切显示;(3)左侧上颌窦炎。
1.2病理学观察
手术后切除标本用10%中性福尔马林固定, 常规取材、制片、HE 染色,显微镜观察。免疫组织化学采用En-Vision二步法,高温高压抗原修复,DAB-H202显色。抗体及检测试剂盒均购自北京中杉金桥公司。
2结果
2.1手术治疗
左侧睑面部及左眼眶内病变切除术。术中见:左侧睑面部13 cm×6.5 cm×2 cm肿块1枚,质韧,表面欠光滑,边界欠清,活动度尚可。
2.2病理检查
2.2.1病理检查 切除皮肤及皮下组织13 cm×6.5 cm×2 cm,皮肤面积11 cm×5.5 cm,皮肤表面粗糙,切面灰白,实性,质中。另见灰褐灰黄不规则组织数块,8 cm×7 cm×2.5 cm,切面灰白,实性,质中。镜下可见神经纤维瘤细胞弥漫分布,呈波浪状排列;丛状神经纤维瘤细胞核呈两端尖细的长梭形,细胞分界不清,呈弯曲波浪状,周围胶原纤维增生,间质中黏液样基质丰富。见图1A、B。病理诊断左侧颜面部皮肤混合性神经纤维瘤,肿瘤成分包括弥漫性神经纤维瘤和丛状神经纤维瘤。
2.2.2免疫组织化学结果 CK(-),S-100弥漫(+),CD34(+),Vimetine(+),bcl(-),SMA(-),Desmine(-),ALK(-),Ki67约2% ,见图1C、D。
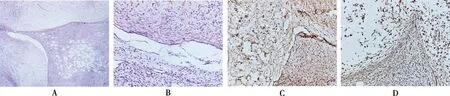
注: A下方为弥漫神经纤维瘤,上方为丛状神经纤维瘤(×20);B上方为丛状神经纤维瘤,下方为弥漫神经纤维瘤,显示细胞呈波浪状排列(×20);C显示S-100弥漫强阳性(×100);D显示vimetine弥漫强阳性(×100)图1 NFⅠ患者病变组织HE染色及S-100、Vimetine免疫组织化学检查Fig.1 HE staining of lesion tissue of NF Ⅰ patient and immuohistochemical staining of S-100 and Vimetine
3讨论
根据美国国立卫生研究所(national institute of health,NIH)1988年提出的神经纤维瘤病的诊断分型标准[2], 包括以下两项或两项以上即可诊断为NFⅠ型:(1)6个或以上的牛奶咖啡斑,青春期前最大直径5 mm以上,青春期后15 mm以上;(2)2个或以上任意类型神经纤维瘤或1个丛状神经纤维瘤;(3)腋窝或腹股沟褐色雀斑;(4)视神经胶质瘤;(5)2个或以上Lisch结节,即虹膜错构瘤;(6)特征性骨损害, 如蝶骨发育不良, 长骨皮质变薄伴或不伴假关节形成;(7)一级亲属(父母、子女和兄弟姐妹)患NFⅠ。本例符合第1、2及第7项,故诊断为家族性NFⅠ型。本例患者的家族史中发病者均为女性,可推测其NF1基因是延母系遗传。相关文献曾报道, NF1基因突变的散发病例,发病率男多于女,而家族性遗传病例,母系遗传多于父系遗传,但整体发病在种族间、性别间无明显差异[3],本例符合此规律。本例患者在家族中的临床表现最重,不仅皮肤有牛奶咖啡斑,颜面部还出现了混合性神经纤维瘤,更进一步证实NF1基因突变率及外显率高的特性。NFⅠ是来源于神经嵴细胞的分化异常,主要是中胚层和外胚层的组织发生改变,可导致多系统损害,属皮肤神经系统损害综合征,患者除以斑块影响面容就诊外,大部分患者都会以神经纤维瘤为首诊,这就提示临床医生在遇到神经纤维瘤病例时,要仔细询问病史,切不可漏诊。本例患者发生的神经纤维瘤是混合性的,在弥漫分布的神经纤维瘤的部分区域出现丛状神经纤维瘤,神经纤维瘤的病理特点是无包膜或包膜不完整,瘤细胞主要为神经鞘细胞,核呈波浪状排列,一般呈两头尖细的长梭形,免疫组化S-100阳性;丛状神经纤维瘤则容易发生在深部,造成外周神经弥漫扭曲和增粗,含大量黏液,瘤细胞常被黏液分离而分界不清。因为神经纤维瘤容易包裹神经而不易切除干净,手术时应综合考虑是否切除神经,应尽可能切除干净,避免复发。丛状神经纤维瘤术后复发率高,恶变可能性比其他类型的神经纤维瘤高约10%[4],临床医生应注意随访。
目前尚无有效的措施能阻止或逆转NFⅠ的病程,只能对其采取对症治疗。曾有报道用中药治疗NFⅠ患者[6],但只是控制病情,并未追踪患者是否治愈。由于NFⅠ可发生骨损害,有研究认为这可能与维生素D缺乏有关,并以此作了相关研究,结果认为补充维生素D以恢复骨代谢是必要的[7]。NFⅠ多表现为牛奶咖啡斑,除部分患者因神经纤维瘤、骨损害等影响了重要器官需要治疗外一般不需处理。
4参考文献
[1] Gottfried ON, Viskochil DH.Molecular,genetic,and cellular athogenesis of neurofibromas and surgicalimplications[J]. Neurosurgery, 2006(58):1-6.
[2] Boyd KP,Gao L,Feng R,et al. Phenotypic variability among café-au-lait macules in neurofibromatosis type 1 [J].Am Acad Dermatol, 2010(3):440-447.
[3] 陈涛.Ⅰ型神经纤维瘤病周围神经病变的超声诊断[J ].中华医学超声杂志, 2012( 10) : 858-860.
[4] Cao X,Xue LF, Sun YM,et al. Nerve-sheath sarcoma with rhabdomyoblastic differentiation(malignant "triton" tumor) at parotid region: case report[J].Journal of Oral and Maxillofacial Surgery Vol, 2015(2):162-164.
[5] 王丹丹,罗阳,桂秋萍,等.结节性硬化9例临床病理特点[J].中华神经科杂志, 2014(3):149-152.
[6] 王俊志,王若馨.中医药治疗皮肤型神经纤维瘤病1例[J].中外医学研究, 2013(27):41
[7] Petramala L, Giustini S, Zinnamosca L,et al. Bone mineral metabolism in patients with neurofibromatosis type 1 (von Recklingausen disease) [J]. Archives of Dermatological Research, 2012(4):325-331.
(2015-10-13收稿,2015-12-29修回)
编辑: 周凌
[中图分类号]R745
[文献标识码]A
[文章编号]1000-2707(2016)01-0121-02
*[基金项目]贵州省科技厅资助项目[黔科合J字(2008)2278号]
**通信作者 E-mail:xushu@gmc.edu.cn
网络出版时间:2016-01-07网络出版地址:http://www.cnki.net/kcms/detail/52.5012.R.20160107.2037.044.html
猜你喜欢
中外医学研究(2016年28期)2016-11-28
中国实用医药(2016年25期)2016-11-03
中国实用医药(2016年9期)2016-05-17
中国实用医药(2016年12期)2016-05-04
中国实用医药(2016年12期)2016-05-04
现代仪器与医疗(2015年6期)2015-12-02
中国现代医生(2015年11期)2015-07-16
中国民族民间医药·下半月(2014年8期)2015-03-11
