
高效液相色谱法测定咳停合剂中吗啡含量
2016-03-27 01:49余孟君欧阳冰卢来春
中国药业 2016年8期
冯 静,余孟君,欧阳冰,卢来春
(中国人民解放军第三军医大学大坪医院野战外科研究所,重庆 400042)
高效液相色谱法测定咳停合剂中吗啡含量
冯 静,余孟君,欧阳冰,卢来春
(中国人民解放军第三军医大学大坪医院野战外科研究所,重庆 400042)
目的 建立测定咳停合剂中吗啡含量的高效液相色谱(HPLC)法。方法 色谱柱为Xtimate C8柱(Welch Xtimate,250 mm×4.6 mm,5 m),流动相为乙腈-0.025 mol/L磷酸二氢钾-0.002 5 mol/L庚烷磺酸钠(30∶85∶85,V/V/V),检测波长为 220 nm,流速为1.0 mL/min,进样量为20 L。结果 吗啡质量浓度在1.008~20.16 g/mL范围内与峰面积呈良好的线性关系,r=0.999 7(n=7),平均回收率为99.16%,RSD=1.28%(n=9)。结论 该方法检测结果准确可靠,灵敏度高,可作为咳停合剂中吗啡的含量测定方法。
高效液相色谱法;咳停合剂;吗啡;含量测定
咳停合剂由复方樟脑酊、远志酊、橙皮酊、氯化铵等组方,具有镇咳、祛痰的功效,适用于支气管炎、上呼吸道感染、支气管扩张及肺炎引起的咳嗽、咯痰。咳停合剂为医院制剂,原质量标准中仅有氯化铵的含量测定和复方樟脑酊[1]的薄层色谱鉴别,不能完全控制咳停合剂的质量。复方樟脑酊为咳停合剂发挥镇咳作用的主要有效成分,且含精神类物质吗啡,会产生依赖性,应对其含量进行控制。为提高咳停合剂质量标准,笔者采用高效液相色谱(HPLC)法对本品复方樟脑酊中的吗啡含量进行了研究[2-6],现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1200型高效液相色谱仪(美国安捷伦科技公司);BAS124S型电子天平(德国Sartorius公司)。
1.2 试药
吗啡对照品(批号为 171201-201324,含量为99.1%)由中国食品药品检定所提供;甲醇、乙腈为色谱纯,试验用水为超纯水,其余试剂均为分析纯;咳停合剂为本院自制(批号分别为 150506-S1,150506-S2,150506-S3)。
2 方法与结果
2.1 色谱条件与系统适用性试验[1]
色谱柱:Welch Xtimate C8柱(250 mm×4.6 mm,5 μm);流动相:乙腈 -0.025 mol/L磷酸二氢钾-0.002 5 mol/L庚烷磺酸钠(30∶85∶85,V/V/V);检测波长:220 nm;流速:1.0 mL/min;进样量:20 μL。
精密量取质量浓度为0.25 mg/mL的吗啡对照品5%醋酸溶液0.5 mL,置处理后的固相萃取柱上,按拟订条件洗脱,用5 mL容量瓶收集洗脱液至刻度,摇匀,作为供试品溶液。分别精密量取供试品溶液与对照品溶液各20 μL,依次注入液相色谱仪,记录色谱图。按下列公式计算,系统适用性测定结果(fs)应在0.97~1.03之间。
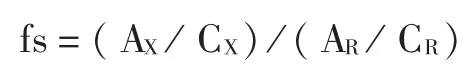
式中,AX为供试品溶液中吗啡峰面积,AR为对照品溶液中吗啡峰面积,CX为供试品溶液质量浓度,CR为对照品溶液质量浓度。
2.2 溶液制备
取固相萃取柱1支,依次用甲醇-水(3∶1)15 mL与水5 mL冲洗,取水适量,滴加氨试液调pH至9,待用。取本品1瓶,超声处理10 min,取出,摇匀;精密量取2 mL,置上述固相柱上,滴加氨试液适量,至柱内溶液的pH约为9(上样前应取同体积上样液预先调试,以确定滴加氨试液的量),摇匀,待溶剂滴尽后,用水20 mL冲洗,用5%醋酸溶液洗脱,用5 mL容量瓶收集洗脱液至刻度,摇匀,作为供试品溶液。取对照品25 mg,精密称定,置100 mL容量瓶中,加5%醋酸溶液溶解并稀释至刻度,摇匀;精密量取1 mL置25 mL容量瓶中,加5%醋酸溶液稀释至刻度,摇匀,作为对照品溶液。称取氯化铵适量,量取远志酊、橙皮酊适量,按处方制成缺复方樟脑酊的咳停合剂,按供试品溶液制备方法制得阴性对照品溶液。
2.3 方法学考察
专属性试验:精密量取供试品溶液、对照品溶液、阴性对照品溶液各20 μL,按拟订色谱条件进样测定。结果,供试品溶液色谱图中,吗啡峰与其他成分峰分离度良好,分离度大于1.5,供试品溶液吗啡峰保留时间与对照品溶液一致,约6.5 min,理论板数大于3 000。色谱图见图1。
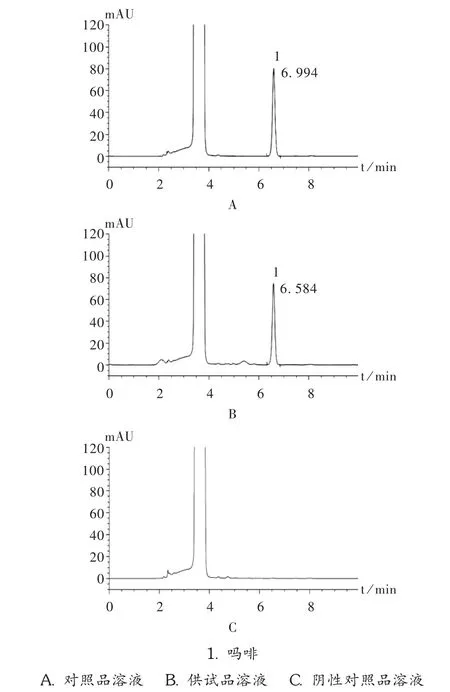
图1 咳停合剂高效液相色谱图
线性关系考察:取吗啡对照品配制系列质量浓度溶液,依法进样测定,以峰面积(A)为纵坐标、质量浓度(C)为横坐标进行线性回归,得回归方程 A=58.708 C+6.014 8,r=0.999 7(n=7)。结果表明,吗啡质量浓度在1.008~20.16 μg/mL范围内与峰面积线性关系良好。
精密度试验:精密吸取同一对照品溶液,连续进样6次,测定。结果的 RSD为0.027%(n=6),表明仪器精密度良好。
定量限考察:精密量取线性关系考察项下对照品溶液,加5%醋酸溶液稀释至0.050 4 μg/mL,按拟订色谱条件进样测定。结果定量限溶液信噪比为12.5,定量限为1.008 ng。
重复性试验:取批号为150506-S1的咳停合剂,依法制备供试品溶液6份,按拟订色谱条件进样测定。结果吗啡的平均含量为 47.40 μg/mL,RSD为 0.18%(n=6),表明该方法重复性良好。
稳定性试验:取重复性试验项下同一供试品溶液,分别于室温下放置0,4,8,14,24 h后,按拟订色谱条件进样测定。结果的 RSD为0.24%(n=5),表明供试品溶液在24 h内稳定性良好。
加样回收试验:精密量取批号为 150506-S1的咳停合剂,加吗啡对照品溶液适量,按拟订方法制备低、中、高3个浓度供试品溶液(80%,100%,120%)各3份,按拟订色谱条件进样测定,计算回收率。结果见表1。

表1 吗啡加样回收试验结果(n=9)
中间精密度试验:取重复性试验项下供试品溶液,放置数天,按拟订色谱条件进样测定。结果2次测得吗啡的平均含量为47.58 μg/mL,RSD为0.78%(n=2),表明该方法的中间精密度良好。
方法耐用性试验:对拟订色谱条件分别进行柱温(±2℃)、流速(±0.2 mL/min)、流动相比例(±2%)的微小改变,精密量取供试品溶液、对照品溶液各20 μL,按拟订色谱条件进样测定。结果咳停合剂色谱条件(流速、柱温和流动相比例)发生微小改变时,吗啡平均含量无明显变化,表明该方法耐用性良好。
2.4 样品含量测定
取3批咳停合剂,依法制备供试品溶液各3份,按拟订色谱条件进样测定,以外标法计算其含量。结果3批供试品溶液中吗啡的含量分别为 47.51,47.68,48.24 μg/mL,RSD分别为 0.04%,0.53%,1.39%(n=3)。
3 讨论
3.1 检测波长选择
取吗啡对照品适量,以甲醇稀释至10 μg/mL,在200~400 nm波长范围内测定最大吸收波长。结果吗啡在214 nm波长处有最大吸收。试验中供试品溶液以5%醋酸溶液作为溶剂,因醋酸在紫外光低波长处有吸收,对色谱图基线有影响,故参考2010年版《中国药典(二部)》[1]中色谱条件,以220 nm作为吗啡的检测波长。
3.2 流动相选择
试验考察了不同流动相对吗啡色谱图的影响。流动相A:乙腈-水;流动相B:乙腈-0.05 mol/L磷酸二氢钾;流动相C:乙腈-0.05 mol/L磷酸二氢钾-0.1%醋酸;流动相D:乙腈-0.025 mol/L磷酸二氢钾-0.002 5 mol/L庚烷磺酸钠。结果流动相为乙腈-0.025 mol/L磷酸二氢钾-0.002 5 mol/L庚烷磺酸钠时,吗啡色谱峰对称因子、峰宽、板数均较理想,因此选择流动相D作为本试验流动相。
3.3 色谱柱选择
试验考察了不同色谱柱对吗啡色谱峰的影响。色谱柱A:DiamonsilC18柱(250mm×4.6mm,5μm);色谱柱B:Welch Xtimate C8柱(250 mm×4.6 mm,5 μm)。结果,当使用色谱柱A时,吗啡色谱峰对称因子为0.75,当使用色谱柱B时,吗啡色谱峰对称因子为0.95,符合要求,故选择Welch Xtimate C8柱(250 mm×4.6 mm,5 μm)作为本试验的色谱柱。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:592.
[2]钟桂香,严 佳,黄爱文,等.高效液相色谱法测定氯化铵甘草口服溶液中吗啡、愈创甘油醚和甘草酸的含量[J].东南国防医药,2014,16(3):229-232.
[3]董海荣,李 君,李俊芳,等.RP-HPLC法测定咳欣康片中吗啡的含量[J].辽宁中医药大学学报,2009,11(11):187-188.
[4]希尔艾力·吐尔逊,罗玉琴,曼尔丹,等.HPLC法测定维药祖卡木胶囊中吗啡的含量[J].中国药房,2009,20(27):2 116-2 118.
[5]王二兵,赵正保,曲婷丽.HPLC测定消炎止咳片中吗啡的含量[J].中国实验方剂学杂志,2013,19(15):127-129.
[6]王卫锋,卫伟光.HPLC测定固肠止泻丸中吗啡及盐酸小檗碱的含量[J].中成药,2010,32(6):967-970.
Content Determination of Morphine in Anticough Mixture by HPLC
Feng Jing,Yu Mengjun,Ouyang Bing,Lu Laichun
(Daping Hospital,Institute of Surgery Research,Third Military Medical University,Chongqing,China 400042)
Objective To establish an HPLC method of determining the content of morphine in Anticough Mixture.Methods The HPLC was used with a Welch Xtimate C8column(250 mm ×4.6mm,5 μm),the mobile phase consisted of acetonitril-0.025 mol/L potassium dihydrogen phosphate solution-0.002 5 mol/L sodium heptane-1-sulphonate(30∶85∶85,V/V/V)with a flow rate of 1.0 mL/min,the detective wavelength was 220 nm and the injection volume was 20 μL.Results The morphine showed good linear relationship with the peak area in the range of 1.008-20.16 μg/mL,r=0.999 7(n=7),and the average recovery rate was 99.16%,RSD=1.28%(n=9).Conclusion The method is accurate and high sensitivity,it is suitable for the determination of morphine in Anticough Mixture.
HPLC;Anticough Mixture;morphine;content determination
R284.1;R286.0
A
1006-4931(2016)08-0064-03
2015-08-13)
中国人民解放军军队医疗机构制剂标准提高科研专项课题,项目编号:13ZJZ01-6。
猜你喜欢
介入放射学杂志(2022年10期)2022-11-02
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
科技创新与应用(2021年7期)2021-02-04
医药前沿(2020年23期)2020-12-03
首都食品与医药(2020年1期)2020-10-21
河北果树(2020年2期)2020-05-25
中成药(2018年5期)2018-06-06
中成药(2017年7期)2017-11-22
中国比较医学杂志(2017年10期)2017-01-16
