
高效液相色谱法测定乳粉中维生素K1的改进
2016-02-20 07:30
中国乳品工业 2016年11期
(华测检测认证集团上海华测品标检测技术有限公司,上海 201206)
高效液相色谱法测定乳粉中维生素K1的改进
邵艳,翟立斐,王焕新,柴平海
(华测检测认证集团上海华测品标检测技术有限公司,上海 201206)
建立了固相萃取-高效液相色谱法分析乳品中维生素K1的方法。样品经磷酸盐缓冲液和脂肪酶酶解,正己烷提取和三氧化二铝柱纯化后,以正己烷定容,以Eclipse XDB C8色谱柱(配柱后锌粉衍生柱)分析,溶解了10 mmol氯化锌、5 mmol乙酸钠和5 mmol冰醋酸的甲醇为流动相,在0.8 mL/min的流速下,激发波长243 nm,发射波长:430 nm检测。维生素K1的加标回收率为86.4% ~ 92.6%,变异系数为1.3%~2.4%,线性范围为0.05~10.0 μg/mL,相关系数为0.9994,方法的检出限为10 μg/kg。该方法线性范围广,灵敏度高,净化效果好,可满足实际样品分析的需求。
脂肪酶;固相萃取;高效液相色谱;XDB C8色谱柱;维生素K1
0 引言
维生素K1是一种萘醌衍生物,化学名称为2-甲基-3-(3,7,11,15-四甲基-2-十六碳烯基)-1,4-萘醌。维生素K1能控制血液凝结,因此在临床上将维生素K1广泛应用于凝血酶过低、维生素K1缺乏症、新生儿自然出血症的防止[1]。维生素K1是脂溶性的物质,对热、氧及水分的作用很稳定,但在碱性环境下,受阳光照射会分解[2]。目前维生素K1的检测方法主要有电极法[3]、毛细管电泳法[4]、高效液相色谱法[5-8]。一般维生素K1测定所使用的前处理酶解方法,在液相色谱图中目标峰的前面有一个很大的干扰峰,且出峰时间长。本研究优化前处理方法,采用常规的甲醇溶液为流动相,建立了应用C8色谱柱和锌粉柱后衍生柱对维生素K1的高效液相色谱分析方法。
1 实验
1.1材料与试剂
标准物质:维生素K1(99.0%);甲醇(色谱纯);冰醋酸(色谱纯);正己烷(色谱纯);脂肪酶(酶活力≥700 U/mg);中性氧化铝柱(500 mg/6 mL)其他试剂无水乙醇、氯化锌、氢氧化钠、无水硫酸钠等均为国产分析纯。
1.2仪器与设备
高效液相色谱仪,带FLD检测器;色谱柱(Agilent Eclipse XDB-C8,5 μm 4.6×150 mm);锌粉衍生柱(50 mm×4.6 mm,锌粉50 ~70 μm);高速离心机;DK-S28型电热恒温水浴锅;旋转蒸发仪(EYELA OSB-2100);电子天平(AL204);涡旋混合器(WH-861)。
1.3方法
1.3.1 标准溶液的配制及标准曲线
标准储备液:准确称取50 mg维生素K1于25 mL容量瓶中,用正己烷溶解定容,配制成质量浓度为2 000 μg/mL,-18℃储存备用[6]。根据需要浓度用正己烷稀释成20 μg/mL工作液。
标准曲线配制:分别吸取0.1,0.2,0.5,0.8和1.0 mL标准工作液;用正己烷定容至10 mL,配制成质量浓度分别为0.20,0.40,1.00,1.60和2.00 μg/mL的标准溶液系列。
1.3.2 样品前处理
称取2.5 g奶粉于50 mL塑料离心管中,加入25 mL磷酸盐缓冲液(45 ~50℃,1.39 g磷酸二氢钠和1.28 g磷酸氢二钠溶解定容到500 mL),1.0 g脂肪酶,于37℃±5℃水浴振荡器中过夜。取出样液,加2 mL氢氧化钠溶液(浓度10 mol/L),用50 mL乙醇溶液转移入250 mL分液漏斗中,用100 mL正己烷分两次萃取(必要时加饱和氯化钠溶液去乳化)。合并的有机相真空旋蒸干,用2 mL正己烷溶出,在0℃以下冷冻1 h以上,高速离心后,上清液过中性氧化铝柱,用1.5 mL石油醚淋洗,4 mL正己烷洗脱接收到10 mL试管中(控制流出速度,1滴/3 s)。洗脱液置氮吹仪上于40℃±2℃下吹干后,用1 mL正己烷定容,再经0.22 μm微孔滤膜过滤,供检测。
1.3.3 色谱条件
色谱柱:AgilentEclipseXDB-C8,5μm4.6×150mm,锌柱(50 mm×4.6 mm);流速为0.8 mL/min;进样量为10 μL;柱温为30℃;流动相为甲醇溶液(10 mmol氯化锌、5 mmol乙酸钠和5 mmol冰醋酸);荧光检测波长为激发波长243 nm,发射波长430 nm。
2 结果与分析
2.1前处理条件的选择
实验中发现用脂肪酶酶解时,加入磷酸盐缓冲液[9]后,目标峰前面的干扰峰变小。有文献介绍[10],胰脂肪酶的酶解效果不错,但是价格昂贵,出于对检测成本考虑,本研究还是采用脂肪酶酶解,然后再用氧化铝柱纯化,干扰峰明显变小。
2.2色谱柱的选择
一般文献资料里介绍都采用C18柱进行维生素K1的测定。由于维生素K1侧链上有四个异戊二烯残基,所以在C18柱上保留强,导致出峰时间很长,因此采用了C8柱进行了优化。维生素K1在Eclipse XDB-C8色谱柱上的保留时间在6 min左右,而以同样的流速和甲醇溶液流动相,维生素K1在Eclipse XDB-C18色谱柱上的保留时间在15 min左右,峰型也不好,应用C8柱大大缩短了分析检测时间。
2.3流动相和流速的选择
GB 5413.10-2010中测定维生素K1,流动相中加入的二氯甲烷,大量长期使用对仪器有损害,本实验只用甲醇溶液做流动相进行色谱分析,保留时间为5.8 min,流速0.8 mL/min时达到了合适的保留时间、峰型以及分离效果.
2.4绘制标准曲线
标准系列各进样10 μL,得到维生素K1的色谱图,以标准溶液浓度为横坐标,峰面积为纵坐标绘制标准曲线,并对曲线进行线性回归。该方法在质量浓度为0.05 ~10.0 μg/mL范围内线性关系良好。回归方程及相关系数如表1所示。

表1 维生素K1的回归方程和相关系数
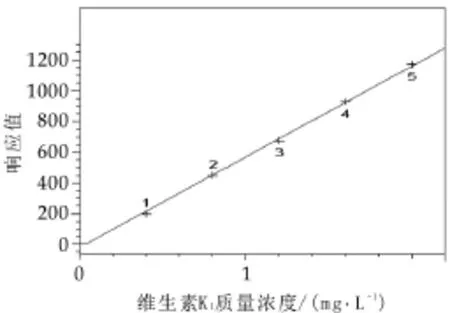
图1 标准曲线
2.5样品分析和检出限
将标准品、加标样品按照方法进行测定,得到其色谱图(图2和图3)。样品的检出限为10 μg/kg。
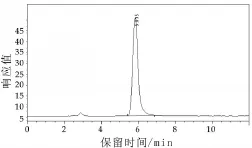
图2 标准色谱
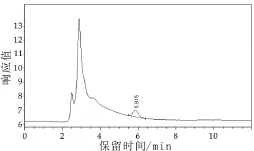
图3 加标样品色谱
2.6回收率、精密度
在奶粉中分别加入10,20,100 μg/kg三个不同质量分数的标准品;分别进行6次平行实验,得到方法的回收率和精密度如表2所示。
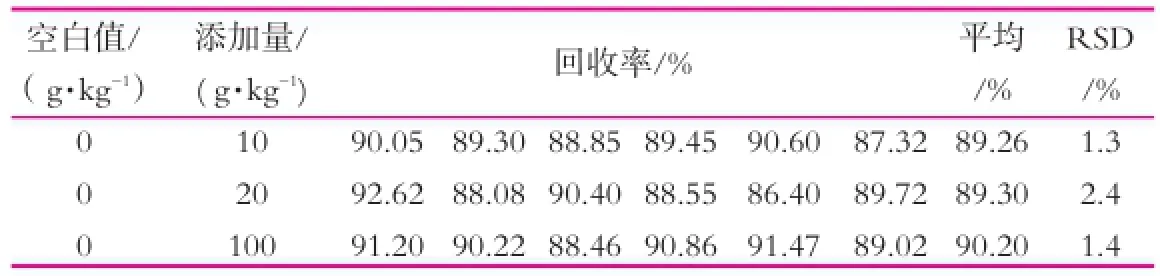
表2 方法回收率和精密度(n=6)
3 结 论
本文建立了高效液相色谱法测定乳粉中的维生素K1。采用固相萃取柱净化,Eclipse XDB C8柱进行高效液相色谱分离,荧光检测器测定。实验结果表明,采用脂肪酶酶解后氧化铝柱纯化,对于乳粉有较好的净化效果,应用C8柱大大缩短了样品检测的时间。实验证明,该方法快捷,结果准确,适用于实验室对乳粉中维生素K1的检测。
[1]王昂,王丽丽,蒋沁,等.维生素K的生产及应用研究[J].河北化工,2008,31(9):35-37.
[2]聂洪勇.维生素及其分析方法[M].上海:科学技术文献出版社,1987.
[3]陈松林,韩紫岩,丁彦庭.修饰电极技术在维生素K1含量测定中的应用[J].河南大学学报,2003,22(3):28-29.
[4]梁红艳,温洁,姜晓峰.利用毛细管电泳测定血清中维生素K的含量[J].中国实验诊断学,2003,7(2):87-89
[5]HARVY E,DAVID C.W.Determination of Vitamin K in Milk and Infant Formulas by Liquid Chromatography;Collaborative Study[J].J. Aoac.Int.,2000,83(1):121-130.
[6]HIROSHI I.Determination of vitamin K1in emulsified nutritional supplements by solid-phase extraction and high-performance liquid chromatography with postcolumn reduction on a platinum catalyst and fluorescence detection[J].J.chromatogra.A,2008,881(1-2):261-266.
[7]GB 5413.10-2010婴幼儿食品和乳品中维生素K1的测定[S].
[8]王竹,王光亚.蔬菜中维生素K1的测定-HPLC法[J].Acta Nutri⁃menta Sinica,1999,21(4):450-453.
[9]邓峰,周英田,吴小红.高效液相色谱法对食品中维生素K1的定量测定[J].色谱,1996,14(3):233-234.
[10]姜瑞清,周琳,胡桂林,等.对GB 5413.10-2010中维生素K1检测的改进研究[J].中国乳品工业,2011,39(9):50-52.
Improvement research on the determination of Vitamin K1in milk using High Performance Liquid Chromatography
SHAO Yan,ZHAI Li-fei,WANG Huan-xin,CHAI Ping-hai
(Shanghai PinBiao Company of Centre Testing International(shenzhen)Corporation,Shanghai 201206,China)
An analytical method was developed for the determination of Vitamin K1in milk by solid phase extraction-high performance liq⁃uid chromatography.The sample was enzymatic hydrolysis by lipase in phosphate buffer solution,extracted with hexane,and then the extract was purified with aluminium oxide column,and finally,Vitamin K1was stored in hexane solution.The separation was achieved by using Eclipse XDB C8 column and an elution with the mobile phases of methanol dissolving 10 mmol Zinc chloride,5 mmol sodium acetate and 5 mmol glacial acetic acid.The identification and quantitative analysis were carried out with fluorescence detector at excitation 243nm and emission 430 nm with flow rate 0.8 mL/min.The recoveries ranged from 86.4%to 92.6%with the relative standard deviations(RSDs)be⁃tween 1.3%and 2.4%.The linear range was 0.05 ~10.0 μg/mL and the correlation coefficient(r2)was 0.9994.The limit of detection was 10 μg/kg.The method is enough for the analysis of Vitamin K1in actual samples with wide linear range and high sensitivity.
lipase;solid phase extraction;High Performance Liquid Chromatography spectrometry;XDB C8 chromatography column;Vitamin K1
TS252.7
A
1001-2230(2016)11-0057-02
2016-03-30
邵艳(1975-),女,硕士,研究方向为食品检测分析。
猜你喜欢
化工设计(2022年4期)2023-01-02
石油炼制与化工(2022年2期)2022-02-15
食品安全导刊(2021年21期)2021-08-30
化工管理(2020年26期)2020-10-09
中国乳业(2020年12期)2020-04-12
广西科技大学学报(2018年2期)2018-09-10
中国洗涤用品工业(2017年2期)2017-04-16
电子技术与软件工程(2016年24期)2017-02-23
食品安全导刊(2014年8期)2014-10-21
中国粮油学报(2014年7期)2014-02-06
