
高效液相色谱法测定动物性食品中马波沙星残留量的研究
2016-02-08 01:24:22贾国宾苏然平刘欣魏丽娟瞿红颖宋婷婷耿智霞贾兴魏占勇黄显会
中国兽药杂志 2016年8期
贾国宾,苏然平,刘欣,魏丽娟*,瞿红颖,宋婷婷,耿智霞,贾兴,魏占勇,黄显会
(1.河北远征药业有限公司,石家庄050041;2.河北省兽药工程技术研究中心,石家庄050041;3.华南农业大学,广州510642)
高效液相色谱法测定动物性食品中马波沙星残留量的研究
贾国宾1,2,苏然平1,2,刘欣1,2,魏丽娟1,2*,瞿红颖1,2,宋婷婷1,2,耿智霞1,2,贾兴1,2,魏占勇1,2,黄显会3
(1.河北远征药业有限公司,石家庄050041;2.河北省兽药工程技术研究中心,石家庄050041;3.华南农业大学,广州510642)
建立了高效液相色谱法测定猪肝脏、肌肉、肾脏、脂肪等组织中马波沙星残留量的方法。以氧氟沙星为内标物,将经过前处理的样品用高效液相色谱仪测定。经验证,马波沙星在肝脏、肌肉等组织中的浓度在0.02~10 μg/g范围内呈良好的线性关系,r≥0.999;方法检出限为0.01 μg/g,最低定量限为0.02 μg/g;在动物组织中以不同水平添加马波沙星,其回收率均在70%~120%之间,批内变异系数≤10%,批间变异系数≤15%。该残留检测方法具有灵敏度高、准确度及精密度高等优点,满足动物性食品安全检测的要求。
高效液相色谱法;动物性食品;马波沙星;残留量
马波沙星(Marbofloxacin)是一种动物专用的新型氟喹诺酮类抗菌药,由瑞士罗氏公司研制。该药在1995年首次在英国上市,先后被英国、法国、美国和欧洲批准用于牛呼吸道感染和大肠杆菌引起的急性乳房炎、猪的无乳综合症,以及犬、猫的胃肠道感染、呼吸道感染和泌尿道感染,具有抗菌谱广,抗菌活性强的特征[1-3]。马波沙星在动物体内分布广泛,耐受性良好,与其他抗菌药物无交叉耐药性等特征,且其生物利用度高,毒性低,被列为动物专用药[4-6]。
近年来,随着氟喹诺酮类药物的普遍使用,其残留检测技术已被国内外广泛研究,目前的检测方法主要包括高效液相色谱法、液相色谱/串联质谱法、毛细管电泳法、酶联免疫法等[7-8],还有许多中外文献报道了同时测定多种氟喹诺酮类药物残留的研究[9-11]。但具体针对马波沙星在动物性食品中的残留检测技术研究较少。马波沙星注射液作为一种新兽药,其研制用于治疗母猪乳腺炎-子宫炎-无乳综合症,为了客观、科学地评价马波沙星注射液在猪体内的残留消除规律,制定合理的临床给药方案,并且为了保证动物性食品的质量安全性,建立了马波沙星在猪组织中残留检测的HPLC方法。
1 材料与方法
1.1 试验设备 高效液相色谱仪(DIONEX Ultimate 3000,戴安);分析天平(AE160,Mettler);离心机(Mach 1.6R,Thermo);旋涡混合仪(MSI,IKA);多功能振荡器(HS250 Basic,IKA);氮气浓缩仪(TurboVap LV,Zymark);可调微量移液器(Eppendorf Research,EPPENDORF)。
1.2 试剂 马波沙星对照品(批号为Y0000819,购自欧洲药典委员会);乙腈(色谱纯,Fisher公司);水(符合GB/T 6682规定的二级水);其它试剂均为国产分析纯。
1.3 试液的配制
1.3.1 马波沙星标准溶液 精密称取适量的马波沙星对照品,用适量乙酸溶解,甲醇稀释,制成1000 μg/mL的马波沙星标准贮备溶液;再分别量取适量的马波沙星标准贮备溶液,用50%甲醇水溶液稀释制成浓度系列为0.20、0.50、1.00、3.00、5.00、10.0、20.0、50.0、100 μg/mL的马波沙星标准工作溶液(4℃保存)。1.3.2 氧氟沙星(内标)标准溶液 精密称取适量氧氟沙星标准品,用水溶解并稀释制成500 μg/mL氧氟沙星标准贮备溶液;准确量取适量的氧氟沙星标准贮备溶液,用50%甲醇水溶液稀释制成浓度10 μg/mL氧氟沙星(内标)标准工作液(4℃保存)。
1.4 试验方法
1.4.1 色谱条件 色谱柱:CNWsil C18(4.6 mm×250 mm,5 μm);流动相:乙腈∶2%甲酸∶1%三乙胺∶水=18∶38∶38∶6(V/V/V/V);流速:1 mL/min;检测器:紫外检测器;检测波长:295 nm;柱温:30 ℃;进样量:20 μL。1.4.2 样品处理 肌肉、肾脏、心脏、肺脏、注射部位、皮脂的提取方法:称取1 g(±0.01 g)试样(冷冻试样应解冻至室温),置于50 mL离心管中。加入100 μL内标(氧氟沙星标准工作液,10 μg/mL),静置3 min,加入5 mL三氯甲烷,旋涡1 min,置摇床上300 r/min振荡10 min后在4 ℃下离心。上清转移至15 mL试管内。重复一次,合并上清液。上清液在45 ℃下N2吹干,用1 mL流动相复溶,涡旋,转移上清液,正己烷除脂,离心后取适量溶液经0.22 μm滤膜过滤至样品瓶中,供HPLC测定[12]。
肝脏的提取方法:称取(1±0.01) g试样(冷冻试样应解冻至室温),同上述方法加入内标,静置3 min,加入5 mL磷酸盐缓冲液(pH 7.4),旋涡1 min,振荡10 min,在4℃下10000 r/min离心10 min。上清转移至50 mL离心管内。重复一次,合并上清液。加入适量氯化钠,摇匀,再加入5 mL三氯甲烷,涡旋、振荡、离心,转移有机相至15 mL试管内,重复一次,合并有机相,45 ℃下N2吹干,用1 mL流动相复溶,涡旋,转移上清液,正己烷除脂,离心后取适量溶液过滤,供HPLC测定。
1.4.3 测定法 根据试样溶液中马波沙星残留量情况,进行标准曲线或单点校准。
1.4.4 空白试验 除不加试验药品外,采用相同的方法进行平行操作。1.4.5 灵敏度 方法的灵敏度以检测限和定量限表示。检测限和定量限的测定方法为在空白组织中添加马波沙星系列标准工作液,制成含马波沙星药物浓度分别为0.005、0.01、0.02、0.05、0.10 μg/g的加标试样进行测定,计算信噪比的平均值。以信噪比S/N=3时定为最低检测限LOD,S/N=10时定为最低定量限LOQ(n=5)。
1.4.6 标准曲线和线性范围 在50 mL的离心管中分别称取1 g空白组织,依次加入一系列己知浓度的标准工作液,制成含马波沙星浓度分别为0.02、0.05、0.15、0.3、0.5、1、2、5、10 μg/g、含氧氟沙星(内标)浓度为1 μg/g的组织样品。按照组织样品处理方法处理后,进样分析。以测得的马波沙星的峰面积与氧氟沙星(内标)的峰面积之比(S)与药物浓度(C)作线性回归,求标准曲线的回归方程和线性相关系数。
1.4.7 回收率和变异系数测定 在1 g空白组织中加入马波沙星标准工作液,制成含马波沙星浓度为0.02、0.15、3.00 μg/g的肝脏、肾脏、肌肉组织样品,添加浓度为0.02、0.15、1.00 μg/g的脂肪组织样品。按照样品处理方法处理后,进样分析。每个浓度重复测定5次,以样品中药物峰面积与内标峰面积之比,求得低、中、高3种浓度的回收率,计算各组变异系数(CV%),验证方法的准确度与精密度。
2 结果
2.1 灵敏度 经过试验测定,本方法最低定量限为0.02 μg/g,最低检测限为0.01 μg/g,表明方法的灵敏度良好,满足残留检测的要求。
2.2 标准曲线和线性范围 试验测得结果如表1中所示,在肝脏、肌肉、肾脏和脂肪组织中,马波沙星浓度在0.02~10 μg/g范围内线性关系均良好,各相关系数r≥0.999。
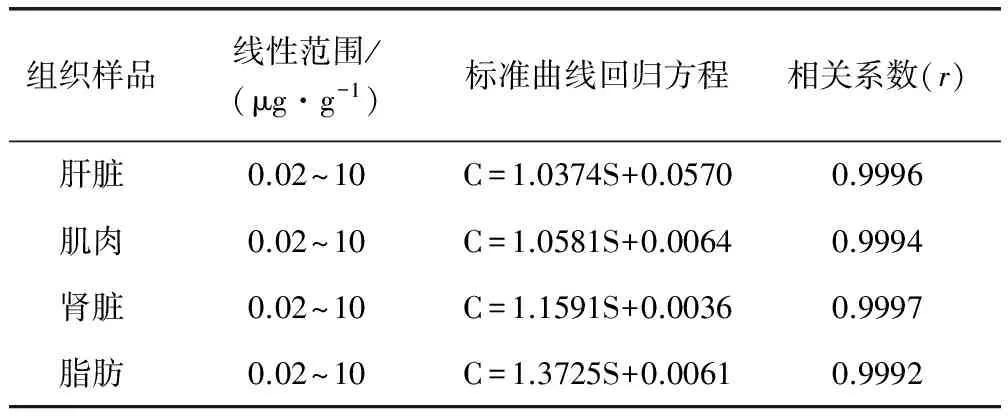
表1 组织样品中添加马波沙星的线性关系
2.3 回收率和变异系数测定 对添加不同浓度马波沙星的组织样品重复测定5次,其测定结果及计算所得的回收率与变异系数如表2至表5所示。由表中数据可知:在猪的肝脏、肌肉、肾脏组织样品中,马波沙星在0.02、0.15、3.00 μg/g三个添加水平上的回收率在70%~120%,批内变异系数CV≤10%,批间变异系数CV≤15%;在猪的脂肪组织样品中,马波沙星在0.02、0.15、1.00 μg/g三个添加水平上的回收率分别在70%~120%,批内、批间变异系数CV≤10%。证明方法的准确度与精密度良好。
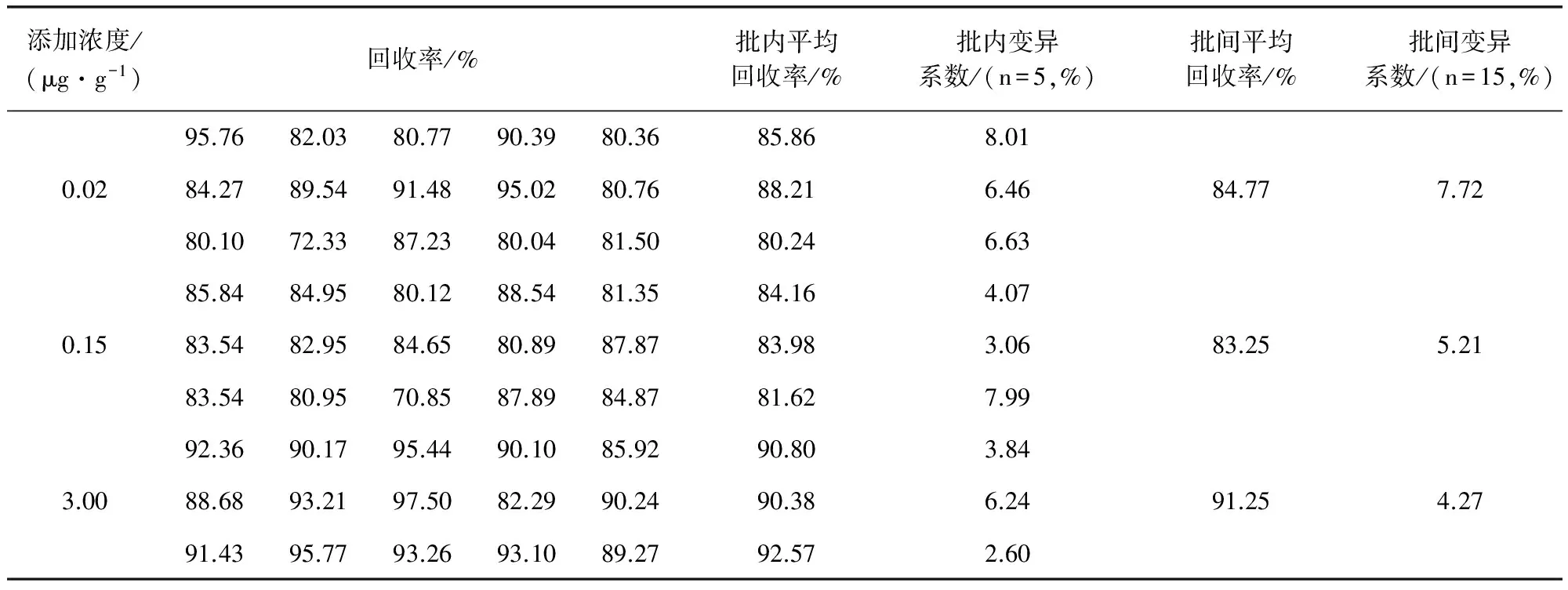
表2 肝脏组织中马波沙星的回收率和变异系数
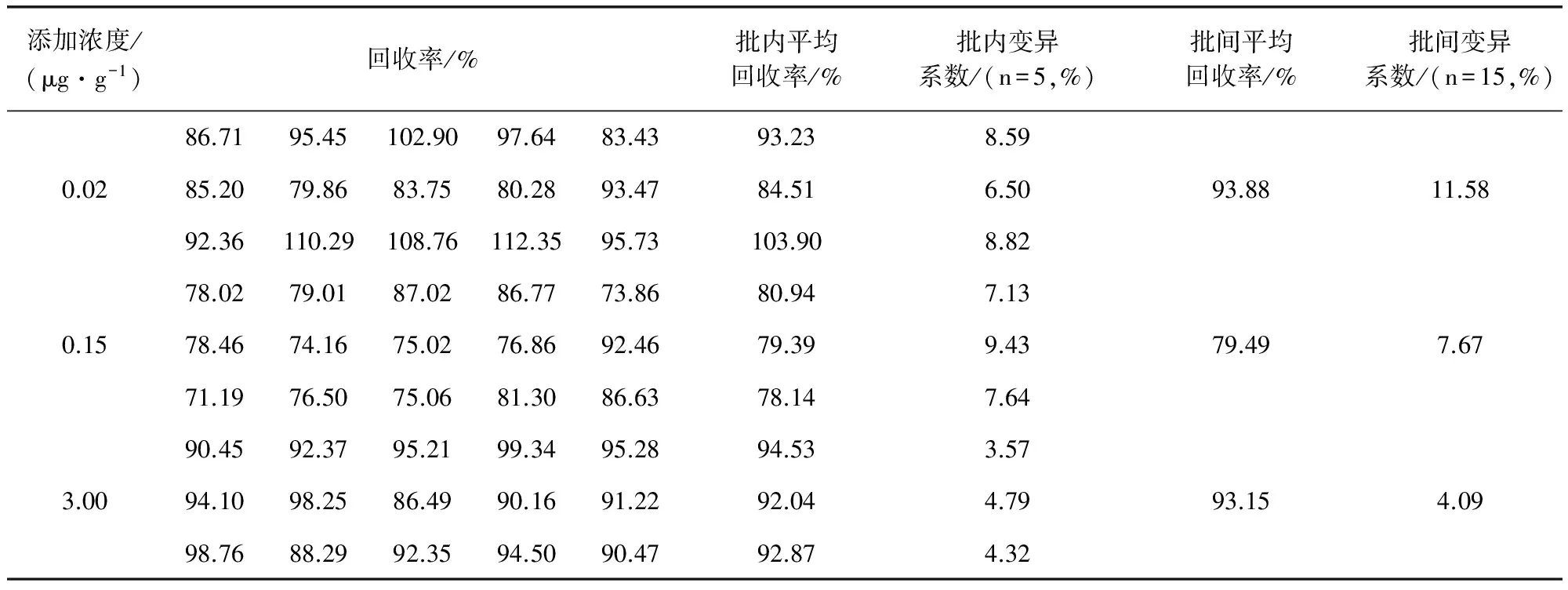
表3 肌肉组织中马波沙星的回收率和变异系数

表4 肾脏组织中马波沙星的回收率和变异系数
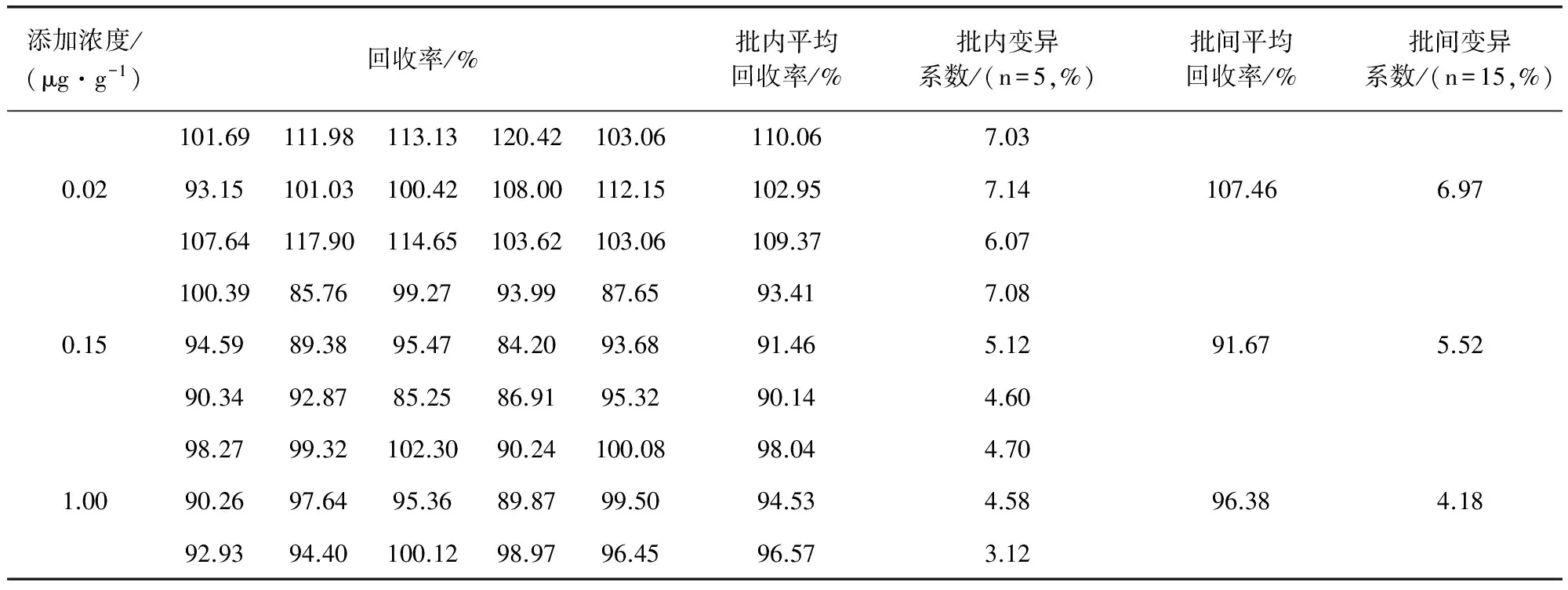
表5 脂肪组织中马波沙星的回收率和变异系数
3 讨论
3.1 样品前处理方法的优化 动物源食品中基质干扰复杂,富含脂肪和蛋白质等多种物质,选择并优化样品的前处理方法对结果的准确性尤为重要。本研究参考文献[12]中的方法进行,对肌肉、肾脏、脂肪等组织样品进行处理,但在提取肝脏样品时发现提取出的样品经过高效液相色谱检测杂质较多,所以采用磷酸盐缓冲液提取并离心取上清液的方法处理,实验发现提取液磷酸盐缓冲液的pH值对肝脏中马波沙星药物残留检测结果影响较大,未经pH调节时,肝脏中残留检测回收率不稳定,当提取液的pH值到7.4时,试验结果较好。所以先用pH 7.4的磷酸盐提取,离心取上清液,再以三氯甲烷萃取,可减少肝脏样品中其他成分对检测的干扰,并且经过检测,此方法测得的样品中马波沙星药物的回收率准确且稳定。
3.2 色谱条件的选择与优化 马波沙星为氟喹诺酮类药物,结构式中含有羧基和哌嗪基,属于酸碱两性化合物,反相液相色谱分析时,色谱峰容易拖尾,通常选择酸性溶液作为流动相的组成部分,同时还加入四丁基溴化铵或三乙胺等扫尾剂以获得好的峰形。本试验尝试了乙腈、乙酸、甲酸、H3PO4、四丁基溴化铵、三乙胺、KH2PO4之间多种配比的流动相条件,经多次比较优化,综合考虑色谱峰的峰值响应、保留时间以及与杂质峰的分离情况,选择乙腈、甲酸与三乙胺的配比作为流动相,选择的比例为乙腈∶2%甲酸∶1%三乙胺∶水=18∶38∶38∶6,此时色谱图基线平稳、峰型良好,内标与马波沙星的保留时间适宜,彼此能够很好的分离且与基质中的杂质峰也能够达到分离要求。
4 结论
本研究针对新兽药马波沙星注射液应用后在动物性食品中的残量检测,建立了高效液相色谱法。该研究具体针对马波沙星注射液使用后,在猪的各组织中的残留检测建立了高效液相色谱方法。建立的方法为内标法,氧氟沙星作为内标物,其加入校准和消除了出于操作条件的波动对分析结果产生的影响,提高了测定结果的准确度。该方法可用于检测猪肌肉、肝脏、肾脏及脂肪等多种组织中马波沙星药物的残留,并且经试验研究验证,方法的灵敏度、准确度与精密度均良好,可用于动物性食品中马波沙星残留量的检测。
[1] 陈军,张淑华. 氟喹诺酮类抗菌药马波沙星的研究进展[J]. 中国兽药杂志,2006,40(12):38-43.
[2] 刘毅. 马波沙星注射液的研制[D].河北医科大学,2015.
[3] Vilalta C, Giboin H, Schneider M. Pharmacokinetic/pharmacodynamic evaluation of marbofloxacin in the treatment of Haemophilus parasuis and Actinobacillus pleuropneumoniae infections in nursery and fattener pigs using Monte Carlo simulations.[J] Vet Pharmacol Ther. 2014,37(6):542-549.
[4] 崔哲. 麻保沙星的药代动力学研究[D].河北科技大学,2011.
[5] 岳永波,杨芳,刘波,等. 马波沙星注射液在母猪体内的药代动力学研究[A]. 中国畜牧兽医学会动物药品学分会.中国畜牧兽医学会动物药品学分会第四届全国会员代表大会暨2011学术年会论文集[C].中国畜牧兽医学会动物药品学分会,2011:101-106.
[6] Schneider M, Paulin A, Dron F,etal. Pharmacokinetics of marbofloxacin in pigs after intravenous and intramuscular administration of a single dose of 8 mg/kg: dose proportionality, influence of the age of the animals and urinary elimination.[J] Vet Pharmacol Ther. 2014,37(6):523-530.
[7] 穆国冬,谭建华. 动物性食品中氟喹诺酮类兽药残留检测技术研究进展[J]. 动物医学进展,2007,28(1):68-72.
[8] 张家禾,孟婷,周作红,等. 动物性食品中氟喹诺酮类药物残留检测方法的研究进展[J]. 中国畜牧兽医,2014,41(5):262-266.
[9] 李雅丽,郝晓蕾,冀宝庆,等. HPLC-ESI-MS/MS测定动物性食品中19种喹诺酮类药物残留的研究[J]. 食品科学,2008,29(8):502-506.
[10]Yorke J C, Froc P. Quantitation of nine quinolones in chicken tissues by high-performance liquid chromatography with fluorescence detection.[J] Chromatography A. 2000,882(1):63-77.
[11]孙雷,朱馨乐,刘琪,等. 猪肉组织中7种氟喹诺酮类药物残留检测高效液相色谱-串联质谱法研究[J]. 中国兽药杂志,2008,42(3):12-15.
[12]李帅鹏,黄显会,孔祥凯,等. 肌注马波沙星在猪组织中残留消除规律[J]. 畜牧兽医学报,2014,45(5):827-832.
(编辑:陈希)
Determination of Marbofloxacin Residues in Animal Derived Food by HPLC
JIA Guo-bin1,2, SU Ran-ping1,2, LIU Xin1,2, WEI Li-juan1,2*, QU Hong-ying1,2, SONG Ting-ting1,2,GENG Zhi-xia1,2, JIA Xing1,2, WEI Zhan-yong1,2, HUANG Xian-hui3
(1.HebeiYuanzhengPharmaceuticalCo.,Ltd.,Shijiazhuang050041,China; 2.HebeiEngineeringResearchCenterofVeterinaryDrugs,Shijiazhuang050041,China; 3.SouthChinaAgriculturalUniversity,Guangzhou510642,China)
A method based on high performance liquid chromatography (HPLC) has been developed for the determination of marbofloxacin in pig liver, muscle, kidney, fat and other tissues. After pretreatment, the tissue samples were analyzed by high performance liquid chromatograph with ofloxacin as an internal standard. The verified results were that: a good linear relationship was obtained for concentration range of 0.02~10 μg/g in the liver, muscle and other tissues (r≥0.999); the detection limit of the method was 0.01 μg/g and the lowest limit of quantification was 0.02 μg/g; in animal tissues, marbofloxacin was added at different levels and the recoveries were at the range of 70%~120%, the intra-assay coefficient of variation was ≤10% and the inter-assay coefficient of variation was ≤15%. The residue detection method had the advantage of high sensitivity, accuracy and precision and it could meet the safety test requirements of animal derived food.
high performance liquid chromatography; animal derived food; marbofloxacin; residues
贾国宾,高级工程师,从事药物制剂方面的研究。
魏丽娟。E-mail:yuanchengcheng@sina.com
2016-01-25
A
1002-1280 (2016) 08-0039-05
S859.84
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:56
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中成药(2018年6期)2018-07-11 03:01:32
大东方(2017年10期)2017-05-30 18:29:24
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:34
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
兽医导刊(2016年12期)2016-05-17 03:51:52
中国粮油学报(2016年5期)2016-01-23 02:45:06
