
Zr和(或)Nb掺杂γ-TiAl合金的延性与结构稳定性
2016-02-08 07:28:34宋庆功杨宝宝赵俊普郭艳蕊胡雪兰
中国民航大学学报 2016年6期
宋庆功,杨宝宝,赵俊普,郭艳蕊,胡雪兰
(中国民航大学a.低维材料与技术研究所;b.中欧航空工程师学院,天津 300300)
Zr和(或)Nb掺杂γ-TiAl合金的延性与结构稳定性
宋庆功a,b,杨宝宝a,赵俊普a,郭艳蕊a,胡雪兰b
(中国民航大学a.低维材料与技术研究所;b.中欧航空工程师学院,天津 300300)
采用密度泛函理论结合物理化学分析,计算了Zr和(或)Nb替位掺杂γ-TiAl形成的8个合金体系的几何结构、能量性质、弹性性质、电子性质和化学键特性。结果表明:各个掺杂体系的总能量和原子平均形成能均为负值,表明其具有较好的稳定性。掺杂体系Ti12Al11Zr、Ti12Al11Nb、Ti11NbAl11Zr和Ti11ZrAl11Nb的轴比都有所改善,更接近于1。对单掺杂体系和双掺杂体系,晶胞体积的变化与杂质原子半径的变化趋势一致,且Zr和Nb总是都倾向于替代Ti原子;掺杂体系均具有较好的延性,为改善γ-TiAl基合金的延性提供了理论依据;Zr和Nb掺杂使体系的共价键强度降低、金属键强度增强,体系的能量稳定性有所降低,因而提高了面间的可动性,有利于改善合金的延性。
γ-TiAl基合金;Zr和(或)Nb掺杂;延性;电子性质;第一性原理
γ-TiAl合金具有较低密度和较高比强度,同时还具有较高的熔点和高温抗蠕变性能等优点,因而成为航空航天、汽车发动机以及工业动力装置中综合性能较好的新型轻质耐热结构材料[1-3]。但由于其室温延性和高温抗氧化性能较差等缺点,给实际应用造成了很大困难[4-5]。为改善γ-TiAl基合金的性能,学界进行了大量理论探索和实验研究[6-10],其中合金化和微合金化方法受到更多青睐。
宋庆功等[11]进行了Zr替位掺杂γ-TiAl基合金的第一性原理研究,发现Zr能够有效改善γ-TiAl基合金的延性与韧性;骆晨等[12]研究了Zr对TiAl合金力学性能在高温时发生的变化,发现添加少量Zr可延长合金的持久寿命;张兰芝等[13]研究发现较低浓度Nb可改善TiAl合金的室温韧性;Gerling等[14]发现,当γ-TiAl基合金中加入Nb时,合金的高温抗氧化性和高温抗蠕变性等性质可得到有效改善。
关于Zr和Nb单掺杂γ-TiAl基合金的理论和实验研究已取得了很大进展,但双掺杂的理论研究还未见报道。因此本文采用第一性原理计算方法,选择合金元素Zr和(或)Nb掺杂γ-TiAl作为研究对象,研究替代掺杂后合金体系的延性和结构稳定性,以期对γ-TiAl基合金的理论研究提供一定依据。
1 结构模型和计算方法
1.1 结构模型
纯γ-TiAl合金属于L10超点阵结构[15]。这种结构可视为由一个纯Ti和一个纯Al的简单四方格子相互套构形成的B2型复式格子,其最小结构单元如图1(a)所示。实验测得纯γ-TiAl晶格参量的典型值为a0= b0=2.837 3 Å,c0=4.059 1 Å[16]。许多研究揭示,微量元素替位掺杂是合金化的主要方式之一,并且对合金性质有重要影响。为使研究对象更加接近实验制备的γ-TiAl基合金样品,本文构建3×2×2的γ-TiAl超胞模型。选择Zr和Nb作为合金化元素,构建8个替位掺杂γ-TiAl基合金体系。对4个单掺杂体系,掺杂的Zr或Nb原子随机替代Al或Ti原子,如图1(b)所示。对4个双掺杂体系,掺杂的Zr与Nb原子尽可能远离并替代非近邻的Al或Ti原子,如图1(c)和图1(d)所示。也就是说,在杂质浓度较低的情况下,不考虑杂质原子的聚集情况。
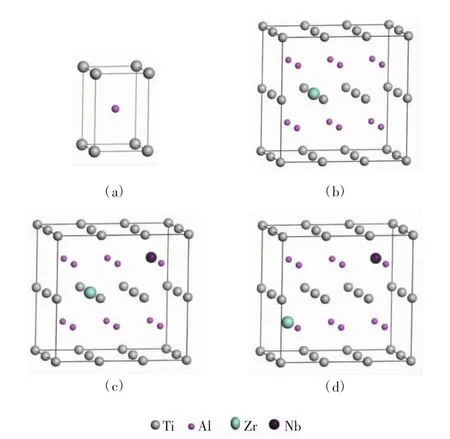
图1 纯的和掺杂的γ-TiA体系的结构模型Fig.1 Structure models of pure and doped γ-TiAl systems
1.2 计算方法与方案
采用密度泛函理论框架下的平面波赝势方法,选择CASTEP(Cambridge sequential total energy package)软件包[17],利用高性能计算集群进行第一性原理计算研究与分析。交换关联函数采用广义梯度近似条件下的PBE形式;电子与离子实之间的相互作用用超软赝势描述。平面波截断能选取为360 eV,k点设置为4× 4×3。自洽场计算采用Pulay密度混合法,自洽计算误差设置为1.0×10-6eV,每个原子能量值低于5.0×10-6eV,每个原子力低于0.1 eV/nm,应力偏差小于0.02 GPa,位移偏移为5.0×10-6A˚。对于各个掺杂体系物理性质的计算研究,均以优化后的晶体几何结构为基础,并采用与该体系的几何优化相同的计算方案来完成。
2 结果与讨论
2.1 掺杂对几何结构的影响
为方便讨论,分别用H0,H1,…,H8表示纯γ-TiAl体系(Ti12Al12),单掺杂和双掺杂γ-TiAl基合金体系如表1所示。几何优化后,各体系的晶格参量也列于表1中。计算结果显示,体系H0的晶格参量为a=8.467 Å,b=5.633 Å,c=8.194 Å,与相对应的实验值(3a0= 8.512 Å,2b0=5.675 Å,2c0=8.118 Å)较为接近,相对误差均在1%以下。表明所选方案比较适合体系H0,对于各掺杂体系的研究,均采用了该计算方案。
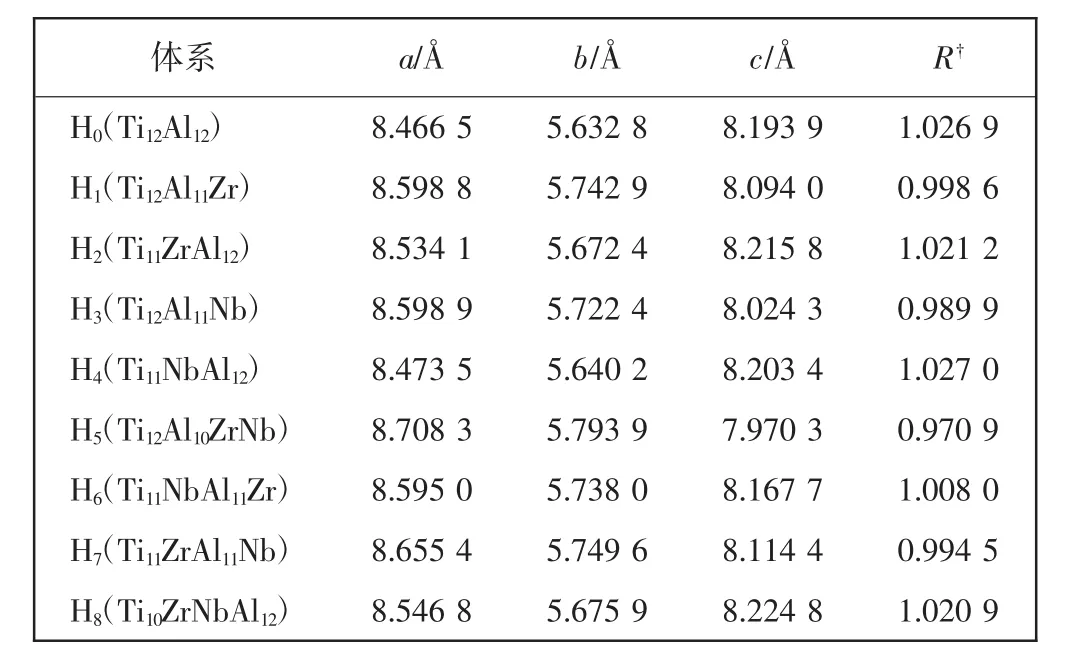
表1 γ-TiAl基合金体系的几何性质Tab.1 Geometrical property of γ-TiAl based alloy systems
由表1可知,Zr、Nb掺杂会使γ-TiAl基合金的晶格参量发生变化而导致其轴比发生改变。Kawabata等[18]研究发现L10结构的γ-TiAl基合金体系的轴比R可以表征其延性的好坏。使轴比c/a减小而接近于1,以提高晶体的对称性和变形协调性并增加滑移系数目,从而使γ-TiAl基合金的延性得以改善。为便于比较,将各体系的轴比绘于图2。由表1和图2可知,掺杂体系H1、H3、H6和H7的轴比均较H0更接近于1。因此,通过适当的替位掺杂可改善此类掺杂体系的立方度,进而使该类合金的延性得以改善。
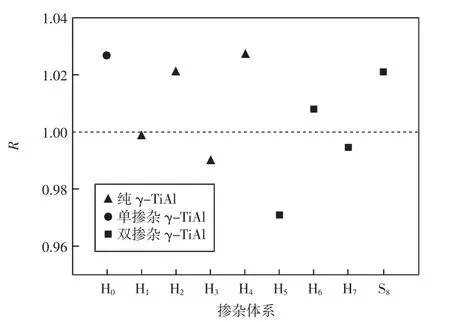
图2 γ-TiAl基合金体系轴比Fig.2 Axis ratios of γ-TiAl based alloy systems
2.2 弹性模量与延性
γ-TiAl基合金属于简单四方晶系,其弹性常数有6个独立分量(C11,C12,C13,C33,C44和C66)。对各稳定体系进行第一性原理计算,得到各体系的弹性常数,如表2所示。
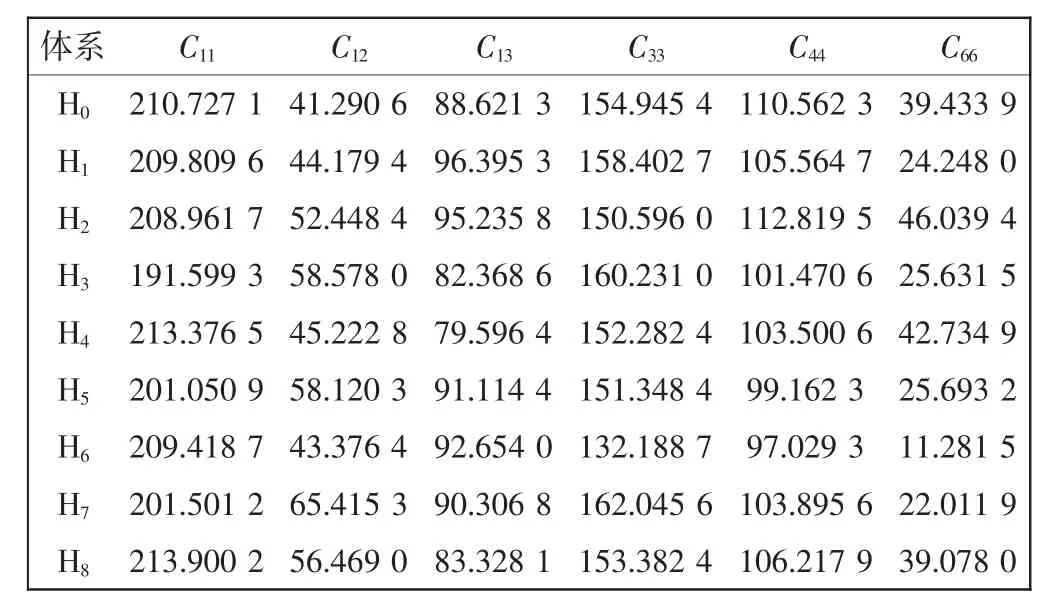
表2 Zr和(或)Nb掺杂γ-TiAl基合金体系的弹性常数Tab.2 Elastic constants of Zr and(or)Nb doped γ-TiAl based alloy systems
四方晶系的体弹性模量B和剪切弹性模量G与弹性常数的关系[19]分别为
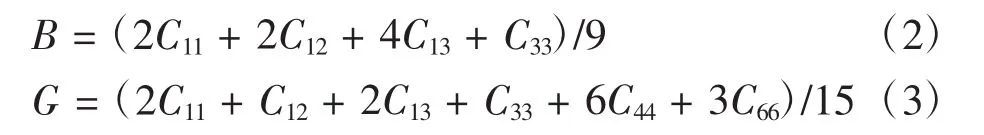
根据式(2)和式(3),计算得到各体系的体弹性模量B和剪切弹性模量G,如表3所示。为便于讨论,计算得到的G/B值也列于表3中。由表3可看出,不同掺杂体系的弹性性质均发生变化。掺杂体系的B值均增大,体系H2、H4和H8的G值增大,其他掺杂的G值均减小。由于B值和G值变化情况各异,因而对体系的延性和塑性产生影响的程度明显不同。
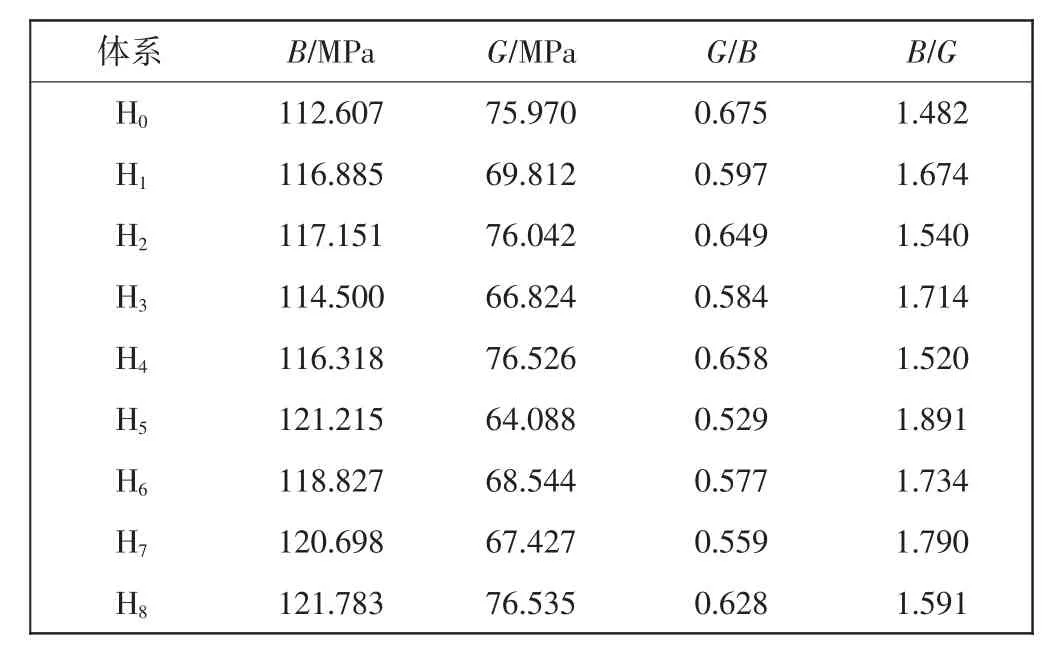
表3 Zr和(或)Nb掺杂γ-TiAl基合金体系的弹性模量及其比值Tab.3 Elastic moduli and ratios of Zr and(or)Nb doped γ-TiAl based alloy systems
Pugh等[20-21]报道,金属间化合物的G/B值可以衡量该类材料的延性。即G/B越大,材料越脆;G/B越小,材料的延性越好。由表3可知,掺杂后所有合金体系的G/B都有所降低,特别是体系H3、H5、H6和H7的G/B值下降明显,对于合金的改善是有利的。其中H3、H6和H7的预测结果与轴比的预测结果一致。而H5的轴比较小,体系的立方度偏离较大,延性尚不能完全定性。
2.3 化学键与延性
掺杂γ-TiAl基合金体系的金属性质与其中原子间结合的化学键密切相关。本文计算了Nb单掺杂体系H3和纯γ-TiAl体系H0中各原子间的重叠布居数,如表4所示。可以看出,在纯γ-TiAl体系H0中,电荷从Ti-4s轨道转移到了Ti-3d轨道,同时也有电荷从Al-3s轨道转移到Al-3p轨道,这直接导致γ-TiAl体系中p-d轨道杂化作用的加强[22]。
纯γ-TiAl中,在平行于ab面的同层Al原子之间有较大的重叠布居数(0.37~0.65),这表明他们之间有很强的共价结合作用;而Al与其近邻Ti原子之间的重叠布居数仅为0.13,也表明有一定的共价键性质。在Nb单掺杂体系(仅以Ti12Al11Nb体系为例)H3中,Nb替代一个Al原子,其与最邻近的8个Ti原子之间的重叠布居数降低为负值(-0.17和-0.16),显示Nb与Ti原子之间形成反键,没有共价结合特征;其它原子间重叠布居数变化不大。结合下文中能带结构的金属特征,推断体系H3中金属键增强,其延性得以改善。
综上所述,Zr和(或)Nb原子替位掺杂能使合金中的共价键强度降低、金属键结合趋势增大,体系稳定性降低,提高了晶面间的可动性,这对合金延性的改善是有利的。这些预测结果与对轴比、弹性模量比的分析结论相一致。
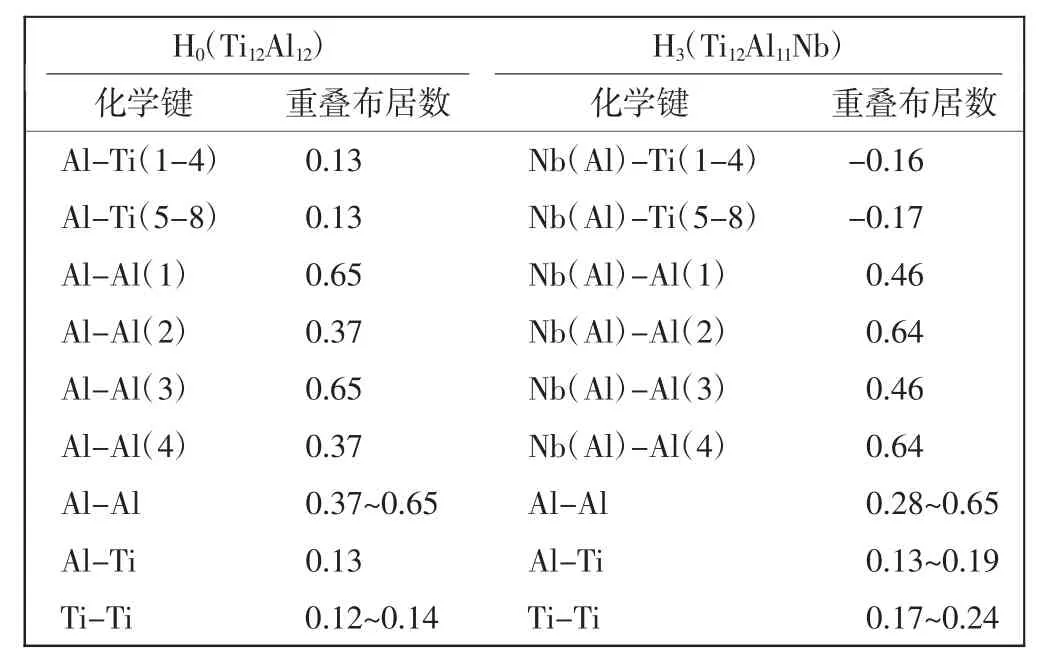
表4 Nb单掺杂与纯γ-TiAl体系中原子间的重叠布居数Tab.4 Overlap population in Nb doped and pure γ-TiAl alloy systems
2.4 形成能和结构稳定性
材料稳定性在一定程度上可用形成能进行表征。原子平均形成能越低,则该材料的稳定性越好[23]。掺杂γ-TiAl基合金体系的原子平均形成能Ef可表示为[24]
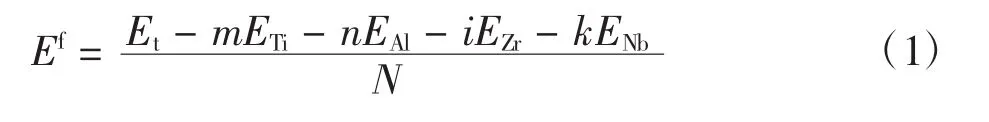
其中:Et为晶胞体系的总能量;m、n、i和k分别表示晶胞中各种元素的原子数;N为晶胞体系的总原子数。ETi、EAl、EZr和ENb分别为Ti、Al、Zr和Nb元素在单质情况下的单原子能量,采用同样的计算方案和参数设置,计算得到其值分别为-1 603.099 9、-56.389 6、-1 281.085 2和-1 551.580 3 eV。计算得到各合金体系的总能量Et,如表5所示。根据式(1)计算各体系的原子平均形成能Ef也列于表5中。
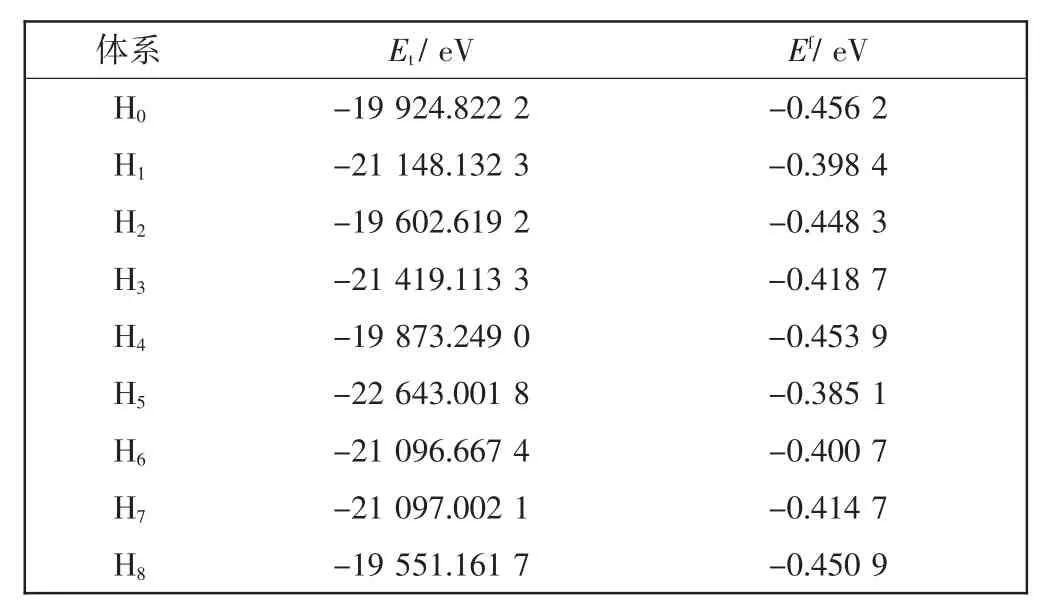
表5 γ-TiAl基合金体系的总能量和形成能Tab.5 Total and formation energies of γ-TiAl based alloy systems
从表5可以看出,Zr和(或)Nb替位掺杂γ-TiAl基合金的总能量和原子平均形成能均为负值,表明这些合金体系都是稳定的。与纯γ-TiAl体系相比,各个掺杂体系的原子平均形成能均有所升高,因而合金体系的稳定性有所降低。对于单掺杂体系说明Zr和Nb原子均倾向于替代Ti,这与Morinaga等[25-27]报道的实验结果可以相互佐证。当Zr和Nb双掺杂时说明4个双掺杂体系的稳定性关系是H8>H7>H6>H5。即双掺杂时,Zr和Nb原子均替代Ti原子的概率较大。上述杂质原子优先占位结论,可由固溶理论解释,已知Ti、Al、Zr和Nb原子半径的关系为rZr>rNb>rTi>rAl[28]。这就使得Zr和Nb替代Al原子的晶格畸变能大于替代Ti原子的晶格畸变能。
体系的负形成能显示其稳定性,也预示着该体系在实验上可以制备。因此,本文预测上述Zr和(或)Nb掺杂体系是可以实验制备的。
2.5 电子性质与结构稳定性
晶体材料的电子性质可通过其能带结构(band structure)和态密度(density of states)来分析。本文仅以延性较好的3个掺杂体系H3、H6和H7与H0进行对比分析。计算得到4个体系的能带结构如图3所示,其中费米能级位于能量为0处。可看出3个掺杂体系的价带能级密集,且价带顶均在费米能级之上,表明其都具有典型的电子导电性质,也就是金属导电性。
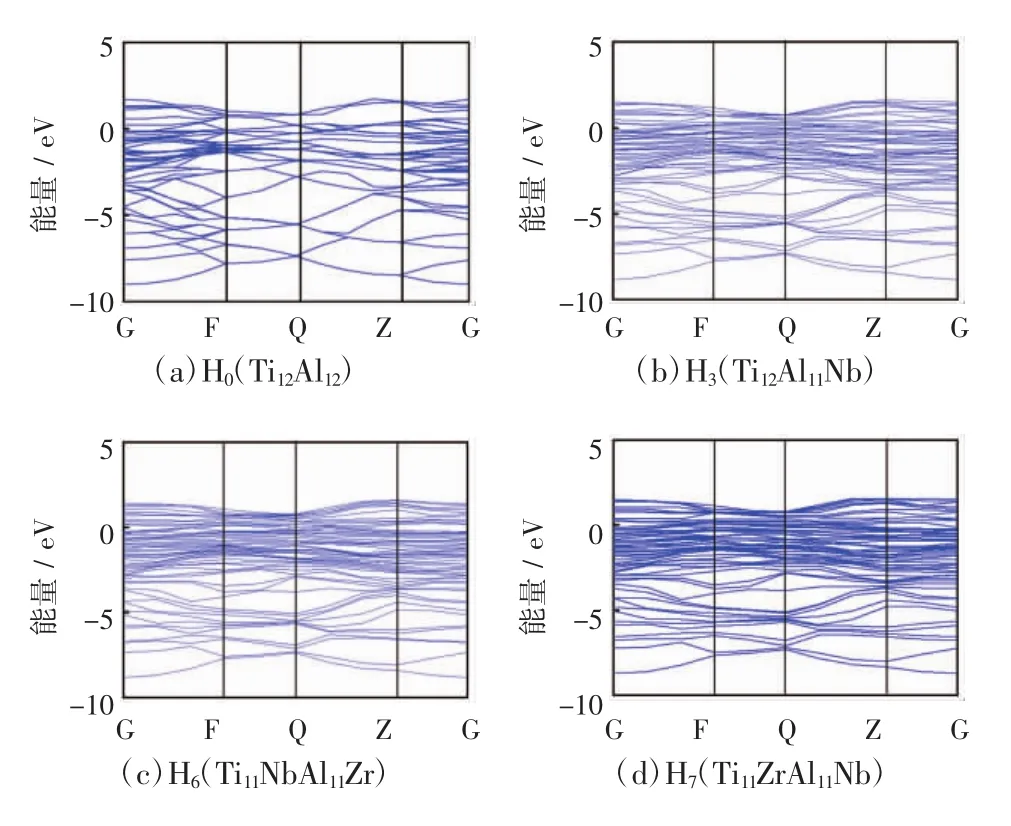
图3 γ-TiAl基合金体系的能带结构Fig.3 Band structures of γ-TiAl based alloy systems
掺杂γ-TiAl基合金体系的电子性质与其结构稳定性也密切相关。Carlsson[29]和Xu[30]研究表明,就不同结构晶体而言,费米能级处的电子态密度越低,其结构越稳定。计算上述4个体系的态密度和分波态密度,如图4所示。具体研究分析体系H0、H3、H6和H7在费米能级处的电子态密度分别为19.158 4、21.831 0、21.352 8和21.517 1 eV。与纯γ-TiAl体系H0相比,3个掺杂体系在费米能级处的态密度均有所增加,表明其结构稳定性均有所降低,与前面依据形成能的定性分析相一致,且体系H6的稳定性最好,H7次之。
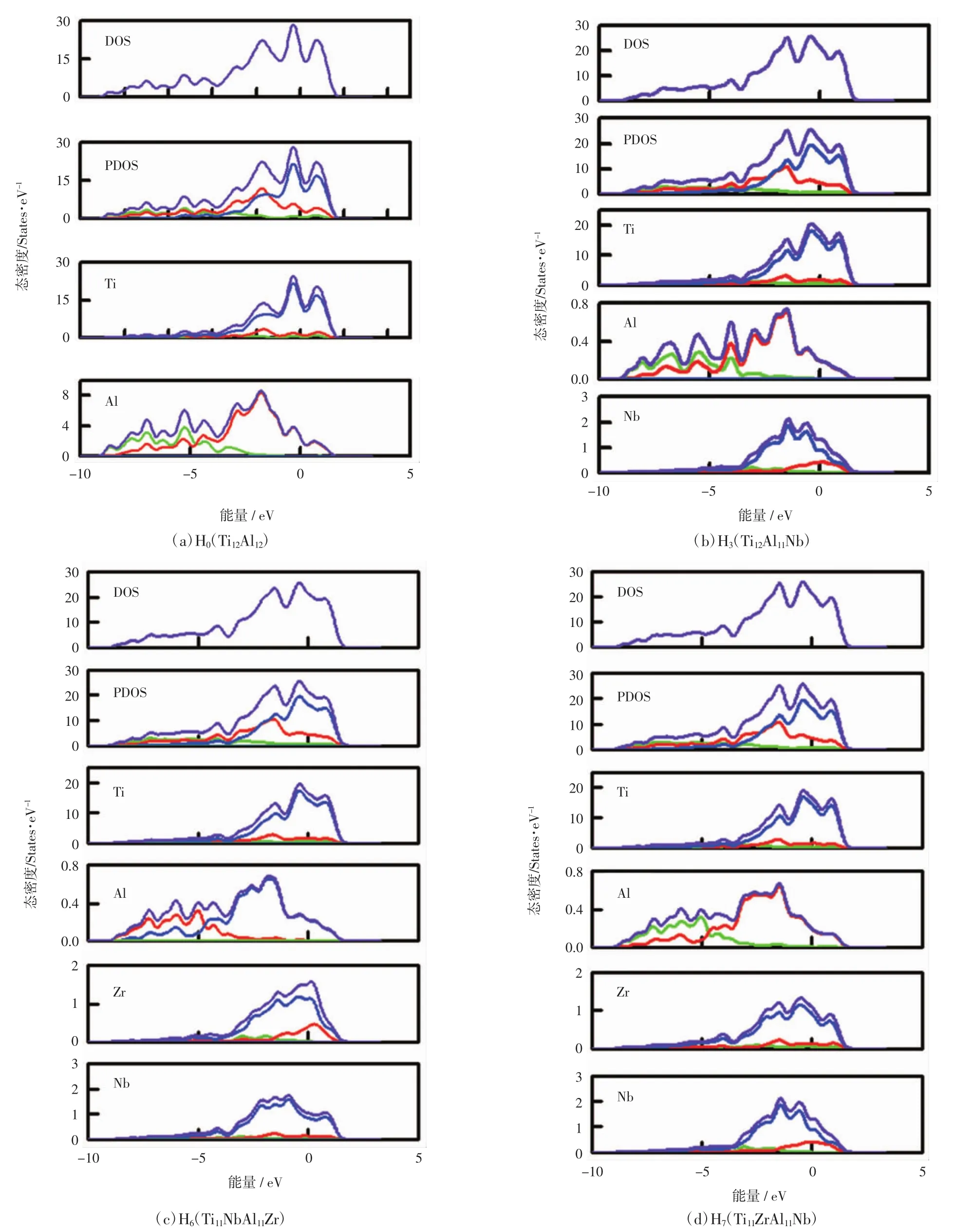
图4 费米能级附近的电子态密度Fig.4 Density of states of systems near Fermi level
3 结语
综合以上对Zr和(或)Nb单(双)替位掺杂γ-TiAl基合金体系的几何结构、形成能与稳定性、弹性与延性、电子性质、重叠布居数的计算与分析,可得以下结论:
1)Zr和(或)Nb单(双)替位掺杂可改善此类γ-TiAl基合金体系的立方度,特别是替位掺杂体系H1(Ti12Al11Zr)、H3(Ti12Al11Nb)、H6(Ti11NbAl11Zr)和H7(Ti11ZrAl11Nb)的轴比都有明显改善,更接近于1。
2)各掺杂体系的总能量和原子平均形成能均为负值,表明其具有较好的稳定性,在实验上是可以制备的。单掺杂情况下,Zr和Nb都倾向于替代Ti原子;双掺杂情况下,Zr和Nb也都倾向于替代Ti原子。
3)延性综合分析揭示,H1、H3、H6和H7轴比均显示其具有较好的延性。根据总能量和形成能的计算结果可知,这4个掺杂体系在实验上均可制备。这为探索改善γ-TiAl基合金的延性提供了依据、揭示了方向。
4)能带结构和态密度显示,3个典型掺杂体系H3、H6和H7都具有典型的金属导电性。经过对原子间重叠布居数的计算与分析,发现用Zr和(或)Nb原子对金属间化合物γ-TiAl进行替位掺杂,能使其中的共价键强度降低、金属键结合趋势增强,提高了晶面间的可动性,有利于合金延性的改善。
[1]KUNAL K,RAMACHANDRAN R,NORMAN M.Advances in gamma titanium aluminides and their manufacturing techniques[J].Progress in Aerospace Sciences,2012,55(12):1-16.
[2]CLEMENS H,MAYER S.Design,processing,microstructure,properties,and applications of advanced intermetallic TiAl alloys[J].Advanced Engineering Materials,2013,15(4):191-215.
[3]HUANG J S,HUANG L,LIU B,et al.Simulation of hot compression of TiAl alloy[J].Intermetallics,2007,15(5/6):700-705.
[4]WU X H.Review of alloy and process development of TiAl alloys[J].Intermetallics,2006,6(10/11):14-22.
[5]陶辉锦,彭 坤,谢佑卿,等.TiAl金属间化合物脆性问题的研究进展[J].粉末冶金材料科学与工程,2007,12(6):330-336.
[6]KARTAVYKH A V,ASNIS E A,PISKUN N V,et al.Microstructure and mechanical properties control of γ-TiAl(Nb,Cr,Zr)intermetallic alloy by induction float zone processing[J].Journal of Alloys and Compounds,2015,643(S1):S182-S186.
[7]IRNAYEV R M,IMAYEV V M,OEHRING M.Alloy design concepts for refined gamma titanium aluminide based alloys[J].Intermetallics, 2007,15(4):451-460.
[8]GOUDA M K,NAKAMURA K,GEPREEL M A H.First-principles study on the effect of alloying elements on the elastic deformation response in beta-titanium alloys[J].Journal of Applied Physics,2015,117 (21):214905(1-6).
[9]SUN F S,CAO C X,YAN M G,et al.Alloying mechanism of beta stabilizers in a TiAl alloy[J].Metallurgical and Materials Transactions,2001, 32(7):1573-1589.
[10]LIU Z C,LIN J P,LI S J,et al.Effects of Nb and Al on the microstructures and mechanical properties of high Nb containing TiAl base alloys [J].Intermetallics,2002,10(7):653-659.
[11]宋庆功,闫洪洋,果福娟,等.Zr替位掺杂γ-TiAl的稳定性和热学性质研究[J].功能材料,2014,45(19):149-154.
[12]骆 晨,吕 楠,朱春雷,等.微量Zr对铸造TiAl合金高温力学性能的影响[J].铸造,2012,61(7):754-757.
[13]张兰芝,王宝义,王丹妮,等.正电子湮没谱研究Nb在TiAl合金中的掺杂效应[J].金属学报,2007,43(3):269-272.
[14]GERLING R,BARTELS A,Clemens H.Structural characterization and tensile properties of a high Nb containing gamma TiAl sheet obtained by powdermetallurgicalprocessing[J].Intermetallics,2004,12(3):275-280.
[15]黄伯云.钛铝基金属间化合物[M].长沙:中南工业大学出版社,1998.
[16]NOVOSELOVA T,MALINOW S,SHA W,et al.High-temperature synchrotron X-ray diffraction study of phases in a gamma TiAl alloy[J]. Materials Science and Engineering A,2004,371(1/2):103-112.
[17]SEGALL M D,LINDAN P J D,PROBERT M J,et al.First-principles simulation:ideas,illustrations and the CASTEP code[J].Journal of Physics:Condensed Matter,2002,14(11):2717-2744.
[18]KAWABATA T,TAMURA T,IZUMI O.Effect of TiAl ratio and Cr,Nb, and Hf additions on material factors and mechanical properties in TiAl [J].Metallurgical and Materials Transactions A,1993,24(1):141-150.
[19]PABST W,GREGOROVA E.Effective elastic properties of aluminazirconia composite ceramics-part 2.Micromechanical modeling[J].Ceram Silik,2004,48(1):14-23.
[20]PUGH S F.Relation between the elastic moduli and the plastic properties of polycrystalline pure metals[J].Philos Mag,1954,45(367):823-843.
[21]FU C L.Electronic,elastic,and fracture properties of trialuminide alloys:Al3Sc and Al3Ti[J].J Mate Res,1990,5(5):971-979.
[22]DANG H L,WANG C Y,YU T.First-principles investigation on alloying effect of Nb and Mo in γ-TiAl[J].Acta physica sinica,2007,56(5): 2838-2844.
[23]宋庆功,姜恩永.快离子导体AgxTiS2中Ag+离子-空位的二维基态结构与能量性质研究[J].物理学报,2008,57(3):1823-1828.
[24]KONG F T,CHEN Z Y,TIAN J,et al.Methods of improving room temperature ductility of TiAl based alloys[J].Rare metal materials and engineering,2003,32(2):81-86.
[25]JIANG C.First-principles study of site occupancy of dilute 3d,4d and 5d transition metal solutes in L10 TiAl[J].Acta Materialia,2008,56 (20):6224-6231.
[26]MORINAGA M,SAITO J,YUKAWA N,et al.Electronic effect on the ductility of alloyed TiAl compound[J].Acta Metal Mater,1990,38(1): 25-29.
[27]HAO Y L,YANG R,CUI Y Y,et al.Estimation of site occupancy error due to statistical noise for the ratio ALCHEMI method[J].Scripta materialia,1999,41(12):1305-1310.
[28]SPEIGHT J G.Lange’s handbook of chemistry[M].16thed.Boston:McGraw-Hill Professional:2005
[29]CARLSSON A E,MESCHTER P J.Relative stability of LI2,DO22,and DO23 structures in MAl3 compounds[J].Journal of Materials Research, 1989,4(5):1060-1063.
[30]XU J H,FREEMAN A J.Band filling and structural stability of cubic trialuminides:YAl3,ZrAl3,and NbAl3[J].Physical Review.Series B, 1989,40(17):11927-11930.
(责任编辑:党亚茹)
Investigations on ductibility of Zr and(or)Nb doped γ-TiAl based alloys
SONG Qinggonga,b,YANG Baobaoa,ZHAO Junpua,GUO Yanruia,HU Xuelanb
(a.Institute of Low Dimension Material and Technology,College of Science; b.Sino-European Institute of Aviation Engineering,CAUC,Tianjin 300300,China)
Eight γ-TiAl based alloy systems doped with Zr and(or)Nb are investigated by using first-principles method based on the density function theory combined with physical chemistry theory.Their geometric structures,energy properties,elastic properties,electronic properties and chemical bond properties are calculated and analyzed. Results indicate that the stability of these systems can be confirmed with their negative total energies and atomic average formation energies.The axis ratios of system Ti12Al11Zr,Ti12Al11Nb,Ti11NbAl11Zr and Ti11ZrAl11Nb are all close to 1,the improvement is distinct.The change tendency of unit volume is in accordance with the radius of doping atom in single-doping and co-doping systems,in addition,Zr and Nb are always inclined to substitute Ti atoms.These doped systems all possess better ductility,presenting theoretical supporting points for the efforts to improve the ductility of γ-TiAl based alloys.It is presented that the doping of Zr and Nb makes the decreased intension of covalent bond while the increased intension of metal bond,resulting in the stability of these alloy decreased slightly.Therefore,it can improve the mobility of crystal face,which is favorable for improving the alloys’ductility.
γ-TiAl based alloy;Zr and(or)Nb doped;ductibility;electronic property;first-principle
TG146.2
:A
:1674-5590(2016)06-0055-06
2016-04-05;
:2016-05-26
:国家自然科学基金项目(51201181);中央高校基本科研业务费专项(3122015K001)
宋庆功(1958—),男,教授,博士,研究方向为新型材料设计与制备.
猜你喜欢
常州工学院学报(2023年5期)2023-11-23 12:09:44
测试技术学报(2022年1期)2022-02-17 06:02:16
现代信息科技(2020年23期)2020-07-09 21:26:25
科技视界(2017年5期)2017-06-30 00:15:19
上海金属(2016年3期)2016-11-23 05:19:43
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
焊接(2016年2期)2016-02-27 13:01:16
汽车文摘(2015年8期)2015-12-15 03:54:08
山东工业技术(2015年2期)2015-07-26 09:03:09
焊接(2015年9期)2015-07-18 11:03:53
