
查尔酮类化合物的抗癌活性研究进展
2016-01-19 00:50王永波,木合布力·阿布力孜
西北药学杂志 2015年3期
关键词:杂环
查尔酮类化合物的抗癌活性研究进展
王永波,木合布力·阿布力孜*
(新疆医科大学药学院药化有机教研室,乌鲁木齐830011)
摘要:目的从传统天然药物中筛选出结构新颖、生物活性较强、安全有效的抗癌先导化合物。 方法通过比较多种含杂环结构查尔酮类化合物及其抗癌机制来进行分析论证。结果在查尔酮结构中,引入不同的基团、改变结构相对构型等使所得化合物的抗癌活性有所提高。结论杂环查尔酮类化合物显示出较强的抗癌生物活性,根据其构型关系,从传统天然药物筛选出结构新颖、生物活性较强、安全有效的抗癌先导化合物。
关键词:查尔酮类化合物;抗癌活性;杂环
doi:10.3969/j.issn.1004-2407.2015.03.034
中图分类号:R914
文献标志码:A
文章编号:1004-2407(2015)03-0325-05
Abstract:ObjectiveTo screen the lead compounds of anti-cancer with the characteristics of novel structure, strong biological activity, safe and effective from the traditional natural medicines. MethodsVarieties of heterocyclic structure and its anticancer mechanism of chalcone were analyzed and discussed. ResultsIn the structure of chalcone, the biological activity of chalcone compounds can be improved by the introduction of different groups, changing the structure of relative configuration and so on. Conclusion
基金项目:国家自然科学基金(编号:81260379);新疆研究生
作者简介:王永波,男,硕士研究生
收稿日期:(2014-10-20)
Research advance of the anti-cancer activity of chalcone compounds
WANG Yongbo,Mourboul ABLISE*(Department of Medicinal and Organic Chemistry,College of Pharmacy; Xinjiang Medical
University,Urumqi 830011, China)
Heterocyclic chalcones have strong anti-cancer biological activity. According to its structure-activitity relationship, lead compounds of anti-cancer,which have novel structure, strong biological activity, safe and effective, could be selected from the traditional natural medicines.
Key words:chalcone compounds;anticancer activity;heterocyclic ring
科研创新项目(编号:XJGRI2014094)
*通信作者:木合布力·阿布力孜,男,博士,教授,博士后(已出站)
癌症是在全世界范围内引起死亡的重要原因之一,最致命的癌症是肺癌、胃癌、肝癌、结肠癌和乳腺癌。
对癌症干预措施来说,化疗是一种有效的治疗方法[1]。但是,临床现有化疗药物因为存在毒性大和耐受性差的缺点,而传统天然药物具有不良反应少和抗癌活性强的特点[2],抗癌药物研究应从相对无毒或者低毒性的天然资源进行筛选,从而得到具有较少的不良反应的药物。
查尔酮、二氢查尔酮是一类广泛分布于大多数植物中的黄酮类和异黄酮类生物合成最重要前体化合物[3],例如,查尔酮是从粉叶蕨(Pityrogrammacalomelano)[4]、明日叶(Angelicakeiskei)[5-6]、绿茶(Camelliasinensis)、红花(CarthamustinctoriusL)[7]和甘草(GlycyrrhizauralensisFisch)[8]等天然药材中分离出的有效化合物;异补骨脂查尔酮来源于天然药材补骨脂(Psoraleacorylifolia)[9-10],其化学结构是1,3-二苯基丙烯酮,由2个芳香环和3个碳原子的α,β-不饱和羰基连接,具有一定生物活性。
1查尔酮衍生物相关生物活性
Sharma等[11]研究表明,吲哚是一类具有重要生物活性、用途广泛的小分子物质。因此,研究者合成了一系列吲哚基查尔酮类化合物,并评价其体外抗癌活性,其中化合物1和2(图1)对PaCa-2细胞系的IC50值分别为0.03和0.09 μmol·L-1[12],被认为是最有潜力和选择性的抗癌药物。

图1化合物1和2
Fig.1 Compounds 1 and 2
Kamal等根据流式细胞仪数据分析细胞周期分布,研究具有咪唑啉酮结构的新型查尔酮抗癌活性。化合物3,4和5(图2)对一组p53人类肿瘤细胞系表现出较好的抗肿瘤活性,这些人类肿瘤细胞系来自9个不同癌症细胞类型,分别是白血病、肺癌、结肠癌、中枢神经系统癌、黑色素瘤、卵巢癌、肾癌、前列腺癌和乳腺癌,它们的GI50值在1.26~13.9 μmol·L-1之间。然而,化合物4和5在浓度为10 μmol·L-1的条件下对乳腺癌细胞(MCF-7)能够引起细胞周期停滞(G2/M期)[13]。

图2化合物3, 4和5
Fig.2 Compounds 3, 4 and 5
Modzelewska等合成一系列含有硼酸基的新型查尔酮和双查尔酮,评价其对人类乳腺癌细胞系MDA-MB-231(雌激素受体阴性)和MCF7(雌激素受体阳性)的抗癌活性,并且与2个正常乳腺上皮细胞系(MCF-10A和MCF-12A)相比较。经过实验证明,含有硼酸基哌嗪结构的双查尔酮6(图3)具有潜在的生物活性。此外,还发现化合物6对结肠癌细胞等位基因HCT116也有细胞毒性,这一对结肠癌细胞等位基因HCT116只是在p53基因表达上不同。与缺乏p53基因的细胞,如与HCT 116 p53-/-(IC50=5.5±0.23 μmol·L-1)相比较,野生型p53细胞(HCT 116 P53+/+)对细胞毒性作用(IC50=2.8±0.32 μmol·L-1)更为敏感,这表明生长抑制依赖于p53基因的表达。分子模型研究已经表明,主要是MDM2癌蛋白赖氨酸残基的K51和K94类似物有效结合[14]。
Sashidhara等[15]合成一系列香豆素类查耳酮,并用一组4种人类癌症细胞系和正常成纤维细胞(NIH3T3)评价它们的体外细胞毒性(SRB法)。在筛选21种化合物中,化合物7(图3)对C33A(子宫颈癌)细胞系比正常纤维细胞NIH3T3细胞(IC50=3.59 μmol·L-1)的选择性大30倍左右。

图3化合物6和7
Fig.3 Compounds 6 and 7
经过证明,在MDA-MB231和MCF-7ADRr细胞系中,槲皮素IC50值分别为3.4和3.9 μmol·L-1,而新型吡啶基查尔酮化合物8的IC50值分别为1.2和2.2 μmol·L-1,这表明化合物8(图4)有较好的抗肿瘤活性。
由于吡啶基团查尔酮存在,延长了G2/M期阻滞和增加DNA碎片,这表明细胞已经凋亡[16]。与槲皮素结构部分相似性,化合物8的抗肿瘤活性也有一些本质的区别:
1)化合物8无调节P-糖蛋白的功能,但槲皮素可以。
2)化合物8无影响活性氧自由基,而槲皮素是一种有效的抗氧化剂。
3)化合物8(大而稀疏的染色质紊乱)引导细胞凋亡形态学模式不同于槲皮素(极度浓缩和破碎染色质)细胞凋亡形态学模式。
4)化合物8对正常组织无毒性,而对肿瘤细胞有选择性毒性,这使其成为一种安全的药物,即使在体内高剂量注射也没有任何严重的不良反应[16]。
在最近的一项研究中,Boumendjel等确定[17],通过筛选一系列查尔酮、吲哚查尔酮9(图4)作为潜在的抗癌药物治疗胶质母细胞瘤,结果如下:
1)化合物9在体外可以降低人类和小鼠的胶质母细胞瘤细胞系的增殖;
2)化合物9能够与秋水仙碱竞争诱导抑制微管蛋白聚合的结合位点;
3)化合物9破坏微管的形成,导致G2/M期细胞周期阻滞和细胞凋亡;
4)化合物9是无外排泵的底物,与长春花碱和紫杉醇形成强烈对比;
5)化合物9可以充当双重抑制剂,因为它能抑制P-糖蛋白(P-gp)和乳腺癌耐药蛋白(BCRP)。
通过与其他抗癌药物比较后,化合物9被认为是一种很有前途的新药[17]。

图4化合物8和9
Fig.4 Compounds 8 and 9
Liu和Go合成具有基本官能团查尔酮(单取代和双取代),并阐述了单取代查尔酮要比双取代或者多取代查尔酮有更好的抗增殖活性;但是,良好的抗增殖活性是与大极性基团、氢键基团的缺失、低HOMO能量和缺电子的β碳有着密切的关系。证据表明,查尔酮结构差异不仅影响生物活性,而且影响作用机制[18]。
评价具有基本官能团的哌啶基查尔酮10和哌嗪基查尔酮11(图5)对人乳腺癌(MCF7)和结肠癌(HCT116)细胞系的抗恶性细胞增殖活性。在MCF7和HCT116中,哌啶基查尔酮10的IC50值分别为2.6±20.5和6.7±2.1 μmol·L-1;哌嗪基查尔酮11的IC50值分别为6.2±0.6和14.1±0.7 μmol·L-1。即化合物10和11是干扰细胞周期进程的抗恶性细胞增殖的有效化合物[18]。

图5化合物10和11
Fig.5 Compounds 10 and 11
化合物12~14(图6)在人类前列腺癌细胞中具有明显生物活性,证实4-氨基喹啉具有不同的生物活性。重要的是,在芳香环中,3或4位用H和X卤素作为取代基可以提高芳香环结构的抗癌活性。关于这一点,需要进一步研究阐明这些衍生物的抗肿瘤机制[19]。
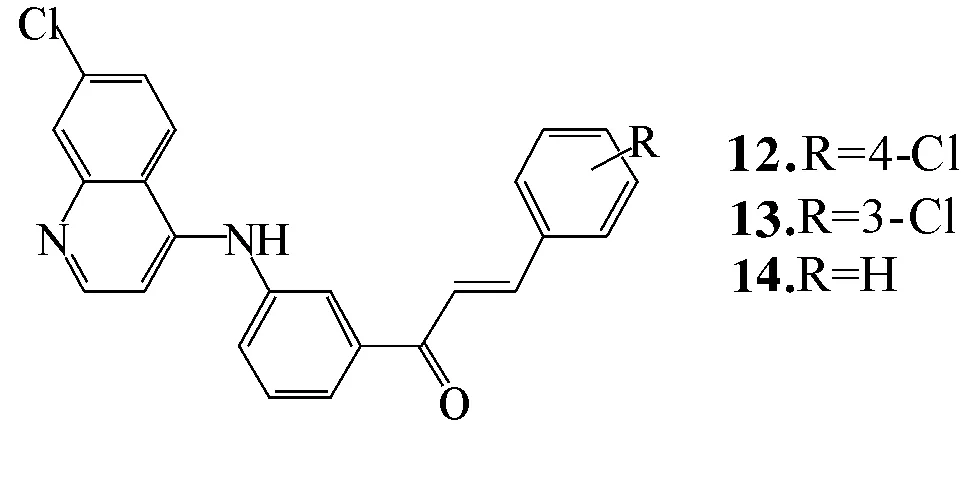
图6化合物12,13和14
Fig.6 Compounds 12,13和14
Romagnoli等制备一些查尔酮的新噻吩类似物,评价其对K562细胞系的抗恶性增殖活性,通过流式细胞仪和分子建模研究显示,它能够与微管蛋白结合引起细胞周期的G2/M期细胞的积累。
在噻吩环5位用邻甲氧基苯基取代,化合物15(图7)通过抑制微管蛋白组装(IC50<2 μmol·L-1)表现出较好的抗恶性细胞增殖的活性。此外,噻吩的双键取代后,仍然保持抗恶性细胞增殖活性以及2个芳香环的构成[20]。
Romagnoli等[21]合成α-溴丙烯酰氨基查尔酮,噻吩环化合物16(图7),具有潜在的抗肿瘤细胞生长的抑制作用。这些化合物在肿瘤细胞中IC50分别是小鼠白血病[L1210;IC50=0.25±0.11 μmol·L-1]、小鼠乳腺癌[FM3A;IC50=0.52±0.06 μmol·L-1]、人体T-淋巴[Molt/4;IC50=0.55±0.18 μmol·L-1]和[CEM;IC50=0.73±0.25 μmol·L-1]和人类宫颈癌[HeLa;IC50=0.34±0.12 μmol·L-1]细胞。因此认为,化合物16迫使细胞进入G0~G1期,导致细胞恶性增殖率降低。

图7 化合物15和16
Fig.7 Compounds 15 and 16
肿瘤细胞的多药耐药性(MDR)是因为外排转运蛋白的超表达,如P-糖蛋白(PGP,ABCB1),MRP1(ABCC1),乳腺癌耐药蛋白(BCRP,ABCG2)[22-25]。
Liu等合成哌嗪基查尔酮类似物,评价在耐药乳腺癌细胞系中耐药性,并得出结论,在合理的浓度范围内,具有二甲基结构的化合物17(图8)能够抑制PGP和BCRP,可能被认为是双重抑制剂[26]。因此,这些抑制剂和抗癌药物共同给药,很有可能作为具有协同作用的抗癌药物。在MCF-7/WT细胞中,化合物17比盐酸多柔比星和米托蒽醌表现出有更大的吸收。

图8化合物17
Fig.8 Compounds 17
2杂环查尔酮衍生物的抗癌机制
药物作用机制可以从不同的角度来看,即作用部位和药物与细胞之间的相互作用。表1总结了各种杂环查尔酮类似物的抗癌作用机制。
表1杂环查尔酮类似物及其抗肿瘤作用机制
Tab.1 Heterocyclic chalcone analogues and anti-cancer mechanisms

杂环查尔酮类似物抗肿瘤作用吲哚解聚微管咪唑阻滞G2/M期哌嗪阻滞G1和G2期吡啶阻滞G2/M期和DNA片段噻吩基抑制TNF-α诱导VCAM-1的表达噻吩抑制微管蛋白装配丙烯酰细胞进入G0~G1峰喹啉抑制拓扑异构酶Ⅱ香豆素抗雄激素
3查尔酮结构与生物活性的重要关系
构效关系是分子化学结构和其生物活性之间的关系。在有机体中,构效关系分析能够测定化学基团引起相应目标生物效应,通过修饰化学结构能够改变具有生物活性化合物的效价。查尔酮的基本结构见图9。

图9 查尔酮基本结构
Fig.9 The basic structure of chalcone
构效关系研究重点:
1)查尔酮衍生物结构简单,易于制备,不饱和酮结构的存在使其显示出良好的生物活性[9];是具有较好应用前景的一类基本骨架结构[27]。
2)在查尔酮A环上,对应用药物生物活性来说,间二甲氧基也是一个不可或缺的部分[28]。
3)结构中用噻吩取代双键之后,仍保持抗增殖活性。因此,改变两个芳环相对构型没有太大的意义[19]。
4)查尔酮S-顺式构象也是影响生物活性的一个至关重要的因素[29]。
5)引入取代基芳香酮可以增强查尔酮类衍生物的药理活性[10]。
6)查尔酮亲脂性对Pgp抑制活性来说是一个非常重要的参数[30-31]。
7)引入氨基能增加查尔酮作为迈克尔受体的反应性,从而提高它们的抗增殖活性[32]。因为在低pH值条件下,氨基基团质子化,增强了吸电子效应。
8)与具有伯氨基的杂环化合物相比,哌啶类查尔酮衍生物很少形成活性代谢物[32-34]。因此,哌啶基查尔酮可能属于杂环抗增殖药物[35]。
9)类异戊二烯链的存在增强了生物膜的相互作用和靶蛋白的亲和力[36-39]。
10)查尔酮的类似物在与DNA相互作用时表现出低的细胞毒作用。与其他化疗药物相比,在很大程度上,查尔酮衍生物消除了诱变性和致癌性的风险[40-41]。
4现状及未来发展方向

在科学研究和工业化生产方面,杂环查尔酮衍生物一直是人们所关注的,但在抗癌活性方面,还需要进一步的研究,不仅应当明确抗癌机制,而且还要激励此类化合物抗癌活性研究。今后,应该重视寻找先导化合物,目的是发现新颖、有效和安全的抗癌候选药物。
参考文献:
[1]Carter S K,Slavik M. Chemotherapy of Cancer[J]. Ann Rev Pharmacol Toxicol, 1974, 14(1):157-183.
[2]Aggarwal B B,Shishodia S.Molecular targets of dietary agents for prevention and therapy of cancer[J]. Biochem Pharmacol, 2006, 71(10):1397-1421.
[3]Star A E, Marby T J. Flavanoid front exudates from two Jamaican ferns,PityrogrammutartareaandP.calomelanos[J]. Phytochemistry, 1971, 10(11):2817-2818.
[4]Sukumaran K,Kuttan R. Screening of 11 ferns of cytotoxic and antitumour potential with special reference toPityrngrummacalomelanos[J]. Ethnopharmacol, 1991, 34(1):93-96.
[5]Akihisa T, Tokuda H, Ukiyaa M, et al. Chalcones, coumarins, and flavanones from the exudates ofAngelicakeiskeiand their chemopreventive effects[J].Cancer Lett, 2003, 201(2):133-137.
[6]Inamori Y,Baba K,Tsujibo H, et al. Antibacterial activity of two chalcones, xanthoangelol and 4-hydroxyderricin, isolated from the root ofAngelicakeiskeiKoidzumi[J].Chem Pharm Bull, 1991, 39(6):1604-1605.
[7]田兰,吴桂荣,王岩.红花黄色素研究进展[J].西北药学杂志,2007,22(4):218-219.
[8]王英华,白虹,窦德强,等.栽培甘草中黄酮类成分的研究[J].西北药学杂志,2004,19(6):253-254.
[9]Rao Y K,Fang S H,Tzeng Y M, et al. Synthesis and biological evaluation of 3′, 4′, 5′-trimethoxychalcone analogues as inhibitors of nitric oxide production and tumor cell proliferation[J]. Bioorg Med Chem, 2009, 17: 7909-7914.
[10]Katsori A M,Hadjipavlou-Litina D. Chalcones in cancer: understanding their role in terms of QSAR[J]. Curr Med Chem, 2009, 16(9):1062-1081.
[11]Sharma V,Kumar P,Pathak D. Biological importance of the indole nucleus in recent years:a comprehensive review[J]. J Het Chem, 2010, 47: 491-502.
[12]Sasajima M,Nakane S,Saziki R, et al. Studies on the anti-ulcer effects of isoprenyl flavonoids (1). The anti-ulcer effects of isoprenyl chalcone extracted fromSophorasubprostrata[J]. Nippon Yakurigaku Zasshi, 1978, 74(8):897-905.
[13]Kamal A,Ramakrishna G,Raju P, et al. Synthesis and anti-cancer activity of chalcone linked imidazolones[J]. Bioorg Med Chem Lett, 2010, 20(16):4865-4869.
[14]Modzelewska A,Pettit C,Achanta G, et al. Anti-cancer activities of novel chalcone and bis-chalcone derivatives[J].Bioorg Med Chem Lett, 2006, 14: 3491-3495.
[15]Sashidhara K V,Kumar A,Kumar M, et al. Synthesis andinvitroevaluation of novel coumarin-chalcone hybrids as potential anticancer agents[J]. Bioorg Med Chem Lett, 2010, 20(24):7205-7211.
[16]De Vincenzo R, Ferlini C,Distefano M, et al.Invitroevaluation of newly developed chalcone analogues in human cancer cells[J]. Cancer Chemother Pharmacol, 2000, 46(4):305-312.
[17]Boumendjel A, McLeer-Florin A,Champelovier P, et al. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP inin-vitroandin-vivoglioblastoma models[J].BMC Cancer, 2009, 9:242.
[18]Liu X,Go M L.Antiproliferative activity of chalcones with basic functionalities[J]. Bioorg Med Chem, 2007, 15:7021-7034.
[19]Ferrer R,Lobo G,Gamboa N, et al. Synthesis of [(7-chloroquinolin-4-yl)amino] chalcones:potential antimalarial and anticancer agents[J]. Sci Pharm, 2009, 77:725-741.
[20]Romagnoli R,Baraldi P G,Carrion M D, et al. Design, synthesis, and biological evaluation of thiophene analogues of chalcones[J]. Bioorg Med Chem, 2008, 16:5367-5376.
[21]Romagnoli R,Baraldi P G, Carrion M D, et al. Hybridα-bromoacryloylamido chalcones. Design, synthesis and biological evaluation[J]. Bioorg Med Chem Lett, 2009, 19(7):2022-2028.
[22]Robert J,Jarry C. Multidrug resistance reversal agents[J].J Med Chem, 2003, 46(23):4805-4817.
[23]Ahmed-Belkacem A,Pozza A,Macalou S, et al. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2)[J]. Anti-cancer Drugs, 2006, 17(3):239-243.
[24]Zamora J M,Pearce H L,Beck W T. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells[J]. Mol Pharmacol, 1988, 33(4):454.
[25]Lee J S,Paull K,Alvarez M, et al. Rhodamine efflux patterns predict P-glycoprotein substrates in the national cancer institute drug screen[J]. Mol Pharmacol, 1994, 46(4):627-638.
[26]Liu X L,Tee H W,Go M L. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein[J]. Bioorg Med Chem, 2008, 16:171-180.
[27]Lawrence N J, McGown A T. The chemistry and biology of antimitotic chalcones and related enone systems[J]. Curr Pharm Des, 2005, 11:1679-1693.
[28]Seelig A,Landwojtowicz E. Structure-activity relationship of P-glycoprotein substrates and modifiers[J]. Eur J Pharm Sci, 2000, 12(1):31-40.
[29]Ducki S,Forrest R,Hadfield J A, et al. Potent antimitotic and cell growth inhibitory properties of substituted chalcones[J]. Bioorg Med Chem Lett, 1998, 8(9):1051-1056.
[30]Bois F,Beney C,Boumendjel A, et al. Halogenated chalcones with high-affinity binding to P-glycoprotein: potential modulators of multidrug resistance[J]. J Med Chem, 1998, 41(21):4161-4164.
[31]Bois F,Boumendjel A,Mariotte A, et al. Synthesis and biological activity of 4-alkoxy chalcones: potential hydrophobic modulators of P-glycoprotein-mediated multidrug resistance[J]. Bioorg Med Chem, 1999, 7(12):2691.
[32]Pati H N,Holt H L,LeBlanc R, et al. Synthesis and cytotoxic properties of nitro- and aminochalcones[J]. Med Chem Res, 2005, 14:19-25.
[33]Xia Y, Yang Z Y,Xia P, et al.Antitumor agents. Part 202: novel 2’-amino chalcones: design, synthesis and biological evaluation[J]. Bioorg Med Chem Lett, 2000, 10(8):699-701.
[34]Dimmock J R,Jha A,Zello G A, et al. Stables[J], J P Pharmazie, 2003, 58: 227.
[35]Liu X, Go M L. Antiproliferative properties of piperidinyl chalcones[J]. Bioorg Med Chem,2006, 14:153-163.
[36]Shimiz K,Kondo R,Sakai K, et al. A geranylated chalcone with 5α-reductase inhibitory properties fromArtocarpusincisus[J]. Phytochemistry, 2000, 54:737-739.
[37]Jayasinghe L,Rupasinghe G K,Hara N, et al. Geranylated phenolic constituents from the fruits ofArtocarpusnobilis[J]. Phytochemistry, 2006, 67(13):1353-1358.
[38]Nishimura R,Tabata K,Arakawa M, et al. Isobavachalcone, a chalcone constituent ofAngelicakeiskei, induces apoptosis in neuroblastoma[J]. Biol Pharm Bull, 2007, 30:1878-1883.
[39]Rodriguez R J,Miranda C L,Stevens J F, et al. Influence of prenylated and non-prenylated flavonoids on liver microsomal lipid peroxidation and oxidative injury in rat hepatocytes[J]. Food Chem Toxicol, 2001, 39:437-445.
[40]Tomas-Barberan F A,Michael N C.Flavones, chalcones and dihydrochalcones-nature, occurrence and dietary burden[J].J Sci Food Agric, 2000, 80(7):1073-1080.
[41]Ivanova Y,Momekov G,Petrov O, et al. Cytotoxic mannich bases of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones[J]. Eur J Med Chem, 2007, 42(11/12):1382-1387.
[42]Kong K W Y, Edler M C,Hamel E, et al. A boronic acid chalcone analog of combretastatin A-4 as a potent anti-proliferation agent[J]. Bioorg Med Chem, 2010, 18:971-977.
[43]Kamal A,Shankaraiah N,Prabhakar S, et al. Solid-phase synthesis of new pyrrolobenzodiazepine-chalcone conjugates: DNA-binding affinity and anticancer activity[J]. Bioorg Med Chem Lett, 2008, 18(7):2434-2439.
猜你喜欢
食品科学(2022年13期)2022-07-29
中国调味品(2022年3期)2022-03-18
河南工业大学学报(自然科学版)(2021年2期)2021-06-01
白城师范学院学报(2021年2期)2021-05-06
科学技术与工程(2020年34期)2021-01-08
世界农药(2019年4期)2019-12-30
肉类研究(2019年8期)2019-09-10
农产品加工(2019年4期)2019-01-06
天然产物研究与开发(2018年10期)2018-11-06
食品安全导刊(2017年15期)2017-02-02
