
糖尿病大鼠肺损伤的发病机制
2015-12-30 09:07周蕾,房辉,田金莉等
中国老年学杂志 2015年8期
糖尿病大鼠肺损伤的发病机制
周蕾房辉田金莉孙雪玲
(唐山市工人医院内分泌二科,河北唐山063000)
摘要〔〕目的观察糖尿病(DM)大鼠肺组织的病理改变及蛋白激酶C(PKC) 、转化生长因子(TGF)-β1、p38丝裂原活化蛋白激酶(p38MAPK)及纤连蛋白(FN)活性的变化,探讨DM大鼠肺纤维化的发病机制。方法实验分为对照组和DM组,并于4、8 w观察血糖、糖化血红蛋白(HbA1c)、体重及肺部组织病理改变,采用免疫组化方法测定PKC、TGF-β1、FN在DM大鼠肺组织表达变化。逆转录聚合酶链反应(RT-PCR)半定量检测肺组织 p38MAPK mRNA的表达。结果与对照组相比较DM组大鼠肺泡间隔及毛细血管壁增宽,肺间质增加,部分肺泡腔萎缩甚至塌陷,肺组织中PKC、TGF-β1、p38 MAPK、FN表达增多、活性增强。结论链脲佐菌素(STZ)糖尿病大鼠肺组织高糖环境下引起血糖、HbA1c升高及体重减轻并引起细胞信号传导系统PKC-TGF-β1-p38MAPK被激活引起FN表达增多从而引起肺组织发生纤维化。
关键词〔〕糖尿病(DM);肺;蛋白激酶C;转化生长因子(TGF)-β1;p38丝裂原活化蛋白激酶;纤连蛋白
中图分类号〔〕R587.1〔文献标识码〕A〔
通讯作者:房辉(1964-),女,博士,教授,主任医师,主要从事内分泌与代谢病研究。
第一作者:周蕾(1982-), 女,硕士,主治医师,主要从事内分泌与代谢病研究。
近年国内外研究〔1〕发现 ,糖尿病(DM)对心、脑、肾有显著的影响。有研究〔2〕表明肺脏也是DM损伤靶器官之一,DM本身可导致肺脏损害,可发展为慢性肺功能障碍〔3〕。DM患者肺功能,可以出现限制性通气功能障碍和弥散、阻塞性及混合性通气功能障碍〔4〕。本实验通过观察DM大鼠肺组织病理学改变及蛋白激酶C(PKC)、转化生长因子(TGF)- β1、纤连蛋白(FN)、p38丝裂原活化蛋白激酶(p38MAPK)的表达的变化,为DM肺损伤的防治提供客观的理论依据。
1材料与方法
1.1动物分组与处理雄性SD大鼠50只。随机分为对照(N)组及糖尿病模型(DM)组,DM组大鼠按60 mg/kg链脲佐菌素(STZ)溶液一次性腹腔注射,N组腹腔注射等量上述的柠檬酸缓冲液,其中20只成模,10只大鼠在饲养过程中因感染、酮症等原因死亡,剔除出本试验,72 h后尾静脉采血测定空腹血糖(FPG)及定性尿糖,FPG≥16.7 mmol/L,尿糖定性≥,动物多饮、多食和尿量增加者确定为DM模型。每组于造模后4、8 w进行指标检测与观察。
1.2标本的收集与处理在禁食12 h后,取大鼠尾末梢毛细血管全血测定FPG。天平称重大鼠体重(BW)。均为1次/w。杀检前1 d空腹尾静脉取血测定糖化血红蛋白(HbA1c)。取上述各组大鼠肺组织行病理学检查及免疫组化实验。
1.3免疫组化肺组织PKC、TGF-β1、FN表达及p38MAPK mRNA表达免疫组化法检测肺组织PKC、TGF-β1、FN免疫组化法检测表达。一抗分别为兔抗鼠PKC、兔抗大鼠TGF -β1、兔抗大鼠FN,二抗为羊抗兔。采用捷达JEDA801D形态学图像分析系统软件,选取染色阳性区域进行计算图像半定量扫描分析。RT-PCR检测大鼠肺组织p38MAPK mRNA的表达。
1.4统计学方法采用SPSS12.0软件进行单因素方差分析。
2结果
2.1各组血糖、HbA1c及BW变化在4、8 w同一时间点,DM组与N组FPG、HbA1c、BW比较有显著差别(P<0.01)。而两组不同时间点之间比较,差别均无统计学意义(P>0.05)。表明在不同病程DM组大鼠均维持在高血糖状态。见表1。
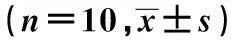
表1 实验动物血糖、HbA1c及
与N组比较:1)P< 0.05;下表同
2.2肺组织病理学变化与N组比较,DM组HE染色结果显示肺泡间隔及毛细血管基底膜增厚,肺间质增加,部分肺泡腔萎缩甚至塌陷,见图1。

图1 4、8 w各组肺组织病理学变化(HE,×40)
2.3免疫组化结果PKC、TGF-β1、FN表达于肺间质及血管上皮细胞核周围的胞质,在肺间质中呈间断不连续分布。N组4、8 w时PKC、TGF-β1、FN蛋白平均光密度值差异均无统计学意义。而DM组4、8 w时PKC、TGF-β1、FN表达的平均光密度值均高于同期N组。不同病程DM组比较,PKC、TGF-β1、FN蛋白表达逐渐增高(P<0.01)。见表2,图2。
表2 实验动物PKC、TGF-β1、FN蛋白表达结果
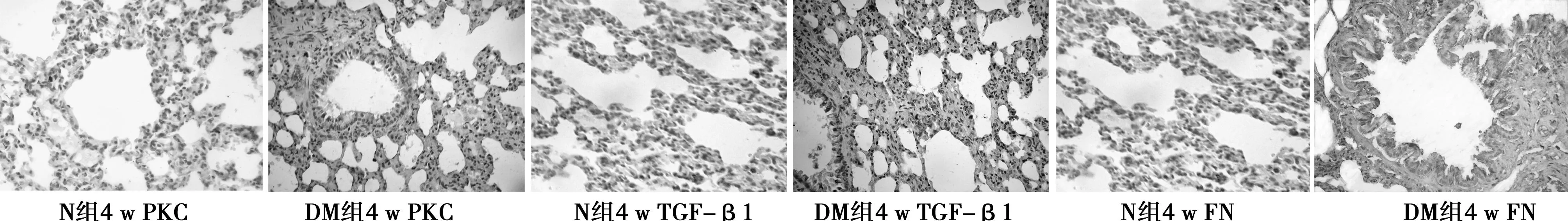
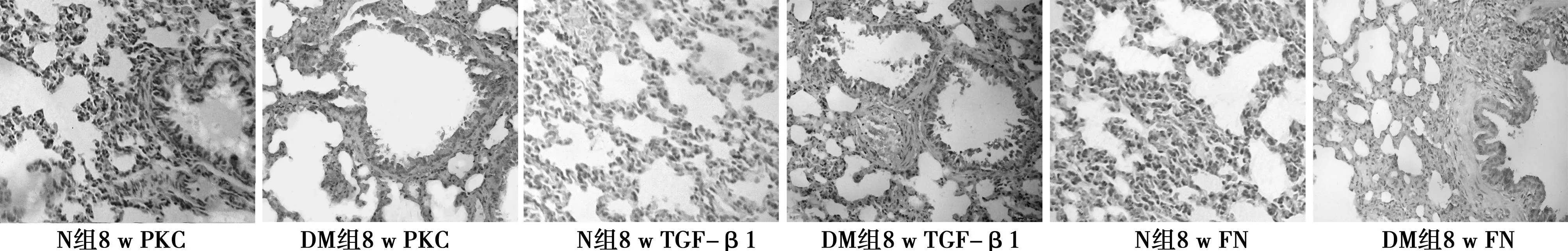
图34、8 w各组肺组织PKC、TGF-β1、FN表达(DAB,×40)
2.4在各组肺组织p38MAPK mRNA表达 与同期N组比较,DM组8 w p38MAPK mRNA表达增高1.68倍(P<0.05),见图3。
1~4:N组4 w、N组8 w、DM组4 w、DM组8 w 图3 p38-MAPK mRNA在肺组织中的表达
3讨论
DM患者基底膜增厚可达 500~800 nm,其基底膜增厚主要涉及肺泡上皮细胞及肺毛细血管内皮细胞的基底膜,可导致肺泡腔缩小〔5〕,肺毛细血管流量下降〔6〕。有关研究〔7〕证实 ,高糖状态下PKC系统参与DM并发症的病理生理过程。PKC主要以非活性形式存在于细胞质中,以活性形式结合于细胞膜。PKC的活化有多条途径,最主要的途径是接受外来信息后产生二酰甘油(DAG),DAG再活化PKC。PKC细胞外调控蛋白激酶通过激活源癌基因复合物c-fos/c-jun二聚体,引起活性蛋白(AP)-1的激活,而AP-1位于TGF-β1基因起动子中。可见PKC激活可引起TGF-β1分泌增加。另外,AP-1还能激活位于纤维黏连素基因启动子区域的环腺苷酸(cAMP)反应原件,从而增强纤维黏连素的表达细胞外基质成分增多。有研究显示报道,TGF-β1通过活化 p38MAPK诱导鼠肾小球系膜细胞的前胶原Ⅰ的合成,提示p38MAPK在 TGF-β1 诱导的ECM合成中起重要作用。ECM重要成分FN的含量增多是促使肺脏纤维化和硬化进行性发展的一个重要因素。FN在细胞增殖和ECM的产生和积聚中发挥重要作用〔8〕。在病理过程中它可以促使成纤维细胞增生并产生更多的ECM,而增多的FN又在各种生长因子的作用下,细胞肥大和纤维细胞增生,这个过程持续发展,ECM不成比例的过度积聚,胶原含量增加,最终导致肺组织纤维化〔9〕。本实验结果提示在早期DM肺损伤过程中PKC蛋白表达显著上调。高血糖状态下PKC引起TGF-β的激活。活化的TGF-β1使p38MAPK进一步磷酸化进入细胞核调控基因表达。刺激肺间质成纤维细胞分泌的FN的表达量明显增加,从而引起DM肺间质成纤维ECM过度分泌,这一过程导致肺间质纤维化。
4参考文献
1Lin CC,Chang CT,Li TC,etal. Objective evidence of impairment of alveolar integrity in patients with non-insulin-dependent diabetes mellitus using radionuclide inhalation lung scan〔J〕.Lung,2002;180(3):181- 6.
2刘松平,张宏. 糖尿病肺损害〔J〕 . 国际内分泌代谢杂志,2004;24(5):341-3.
3Walter RE,Beiser A,Givelber RJ,etal.Association between glycemic state and lung function〔J〕.Am J Respir Crit Care Med,2003;167(6):911-6.
4Marvisi M,Bartonlini L,Borrello P,etal.Pulmonary function in non-insulin-dependent diabetes mellitus〔J〕.Respiration,2001;68(3):268-72.
5Ang LF,Karin M. Mammalian MAP kinase signaling cascades〔J〕.Nature,2001;410(6824):37-40.
6Tsai MH,Shiau YC,Kao CH,etal.Detection of recurrent nasopharyngeal carcinomas after radiotheraphy with 18-fluoro-2-deoxyglucose positron emission tomography and comparison with computed tomography〔J〕.J Cancer Res Oncol,2002;128(5):279-82.
7Matsuoka H,Arai T,Mori M,etal. A p38MAPK inhibitor,FR 2167653,ameliorates murine bleomycin induced pulmonary fibrosis〔J〕.Am J Physiol Lung Cell Mol Physiol,2002;283(1):L103-12.
8Ohshiro Y,Ma RC,Yasuda Y,etal. Reduction of diabetes-induced oxidative stress,fibrotic cytokine expression,and renal dysfunction in protein kinase C beta-null mice〔J〕.Diabete,2006;55(11):3112-20.
9Weynand B,Jonckheere A,Frans A,etal. Diabetes mellitus induces a thickening of the pulmonary basal lamina〔J〕. Respiration,1999; 66 (1):14-9.
〔2013-10-11修回〕
(编辑赵慧玲/曹梦园)
猜你喜欢
听力学及言语疾病杂志(2022年5期)2022-09-20
中国典型病例大全(2022年7期)2022-04-22
科学(2020年2期)2020-08-24
振动与冲击(2017年14期)2017-07-19
中国美容医学(2016年10期)2016-11-29
复旦学报(医学版)(2016年1期)2016-06-20
中国循环杂志(2015年10期)2015-12-24
中国循环杂志(2015年10期)2015-12-24
中华皮肤科杂志(2014年3期)2014-12-19
中国火炬(2014年8期)2014-07-24
