
H2XP…SHY复合物中磷键与硫键的理论研究
2015-12-29 11:18刘玉震黎安勇西南大学化学化工学院重庆400715
物理化学学报 2015年3期
刘玉震 黎安勇(西南大学化学化工学院,重庆400715)
H2XP…SHY复合物中磷键与硫键的理论研究
刘玉震 黎安勇*
(西南大学化学化工学院,重庆400715)
用从头算量子化学方法MP2与CCSD(T)研究了H2XP和SHY(X,Y=H,F,Cl,Br)分子的P与S之间形成的磷键X—P…S与硫键Y—S…P的本质与规律以及取代基X与Y对成键的影响.计算结果表明,硫键比磷键强,连接在Lewis酸上的取代基的电负性增大导致形成的磷键或硫键增强,键能增大,对单体的结构和性质的影响也增大;而连接在Lewis碱上的取代基效应则相反.硫键键能为8.37-23.45 kJ·mol-1,最强的硫键结构是Y电负性最大而X电负性最小的HFS…PH3,CCSD(T)计算的键能是16.04 kJ·mol-1;磷键键能为7.54-14.65 kJ·mol-1,最强的磷键结构是X电负性最大而Y电负性最小的H2FP…SH2,CCSD(T)计算的键能是12.52 kJ·mol-1.对磷键与硫键能量贡献较大的是交换与静电作用.分子间超共轭lp(S)-σ*(PX)与lp(P)-σ*(SY)对磷键与硫键的形成起着重要作用,它导致单体的极化,其中硫键的极化效应较大,从而有一定的共价特征.
σ-hole;磷键;硫键;取代基效应
1 引言
非共价相互作用广泛存在于各种化学和生物体系中,并起着非常重要的作用,1-3它可以改变分子的几何结构、电子结构与性质,以及影响凝聚体系的物理化学性质.氢键是最早发现并得到广泛研究的一种非共价相互作用,4,5随后双氢键6和卤键7-9也先后被发现和深入研究.近年来人们又发现了两种新的非共价相互作用——磷键和硫键,它们分别是P/As或S/Se等第V主族或第VI主族原子作为Lewis酸与其它的富电子原子或基团作为Lewis碱形成的分子间弱相互作用.10-12特别是P、S作为生命体的重要组成元素,它们所形成的非共价键对生物体中核酸、蛋白质等有机大分子的空间结构和生理活性都产生很大影响.
©Editorial office ofActa Physico-Chimica Sinica
所有的非共价相互作用都有一个共同特征:它们都可以看作Lewis酸碱的作用,在Lewis酸中存在正静电势区域,称为σ-hole,而Lewis碱中有负静电势区域,Lewis酸碱正负静电势区域的静电吸引往往是形成非共价相互作用的决定性因素.σ-hole是Politzer13和Murray14等在研究了许多R—X卤键的性质之后首次提出的:由于卤族原子X的pz孤对轨道电子参与形成R—X键,使得X原子pz轨道外层电子缺失,这样就会形成σ-hole,基团R的吸电子能力越强,σ-hole也越强.Scheiner15的工作使得磷键受到重视,他研究了不同取代基X对XP…NH3复合物分子间磷键的影响,并提出强吸电子基X能大大增强P…N磷键的强度,同时他又详细对比和总结了磷键、硫键、卤键和氢键的形成特点与联系.16Del Bene17,18等研究了H2XP…PCX、H2FP…NH2F等复合物中相关磷键和氢键的结构特点与性质.由于吸电子基团X的存在,在P原子附近XP延长线方向上存在一个σ-hole,它的存在就是形成磷键的关键.在磷键之后硫键也受到大量关注,19在SHY分子中,吸电子取代基Y的存在同样使得S原子周围、YS延长线方向存在一个σ-hole,它使得SHY可与Lewis碱形成硫键.
本文中我们研究H2XP和SHY(X,Y=F,Cl,Br)之间形成的磷键和硫键复合物的结构、性质与成键机理.这两种分子中既有σ-hole作为Lewis酸,也有孤对电子作为Lewis碱,因此它们之间可以形成氢键P/X…HS、S/Y…HP,卤键X…S/Y、Y…P/X,磷键X—P…S/Y,以及硫键Y—S…P/X等多种非共价作用,但本文仅研究在P与S之间形成的磷键X—P…S或硫键Y—S…P,考察取代基对该磷键和硫键的影响以及对复合物H2XP…SHY的结构和性质的影响.
2 计算方法
采用二阶微扰理论MP2方法与aug-cc-pVDZ和aug-cc-pVTZ基组对所有单体和复合物进行了构型优化和频率计算,并在aug-cc-pVTZ基组优化构型基础上用CCSD(T)/aug-cc-pVTZ方法做了单点能计算.相互作用能的计算采用了超分子方法,并用Boys和Bernardi提出的Counterpoise方法20做了基组重叠误差(BSSE)矫正.所有的上述计算都是在Gaussian 09软件包21中完成的.电子密度拓扑分析使用了AIMAll软件,22自然键轨道(NBO)分析结果使用了GenNBO 5.0程序.23定域分子轨道能量分解分析(LMOEDA)24采用了GAMESS.64程序.25
3 结果分析与讨论
3.1 磷键与硫键的形成
由于XP与YS延长线处σ-hole的存在,H2XP和SHY可以形成两种P…S相互作用的复合物构型:一种是磷键构型X—Pδ+…Sδ-,H2XP是Lewis酸, SHY为Lewis碱,S提供孤对电子;另一种是硫键构型Y—Sδ+…Pδ-,SHY是Lewis酸,PH2X为Lewis碱, P提供孤对电子.图1中给出了这两类复合物的结构图.未取代的H3P和H2S不能形成P…S复合物,因为除了H原子附近为正静电势外,P与S原子附近都为负的静电势.本文研究了以下几类体系.第一类, H2X—P…SH2(X=F,Cl,Br),P有σ-hole而S没有,只能形成磷键,记为P1,研究磷氢化物的取代基X的改变对磷键的影响.第二类,HY—S…PH3(Y=F,Cl, Br),S有σ-hole而P没有,只能形成硫键,记为S1,研究硫氢化物的取代基Y的改变对硫键的影响.第三类,H2XP…SYH(X,Y=F,Cl,Br),P与S都有σ-hole与孤对电子,可以形成P键与S键.该类研究了三种结构:磷键结构H2XP…SFH(X=F,Cl,Br),研究X取代基的影响并与(1)类比较,记为P2;硫键结构HYS…PFH2(Y=F,Cl,Br),研究Y取代基的影响并与(2)类比较,记为S2;对称结构H2XP…SXH(X=F, Cl,Br),既有磷键也有硫键,比较这两种键,分别记为P3与S3.

图1 复合物的两种构型:磷键与硫键Fig.1 Two types of complexes:pnicogen bond and chalcogen bond
图2中给出了MP2/aug-cc-pVDZ计算下的PH2Cl和SHCl单体分子在0.0004 a.u.等电子密度面的静电势图,图中颜色由红到蓝表示静电势值从负到正逐渐增大.分子静电势(MEP)图形表明PH2Cl和SHCl分子中ClP与ClS延长线上都明显具有一处正静电势区域,这就是σ-hole.用Multiwfn软件26计算的单体分子范德瓦尔斯面上σ-hole区域最大正静电势值:对PH2F、PH2Cl和PH2Br分别为180、159和151 kJ·mol-1;而对SHF、SHCl和SHBr依次为201、151和130 kJ·mol-1.因此,对于PH2X和SHY单体,随着取代卤素原子X、Y从F、Cl、Br的递变,取代基电负性逐渐减小,其σ-hole区域最大静电势值逐渐减小,亲电性依次降低.取代基的吸电子能力越强, σ-hole越强,相应地,其形成的磷键或硫键就越稳定.图2显示PH2Cl与SHCl的静电势分布呈现明显不同,对于相同的取代基X=Y,S的σ-hole强于P的σ-hole,由此可以预计硫键强于对应的磷键,下文的数据支持了这一预言.
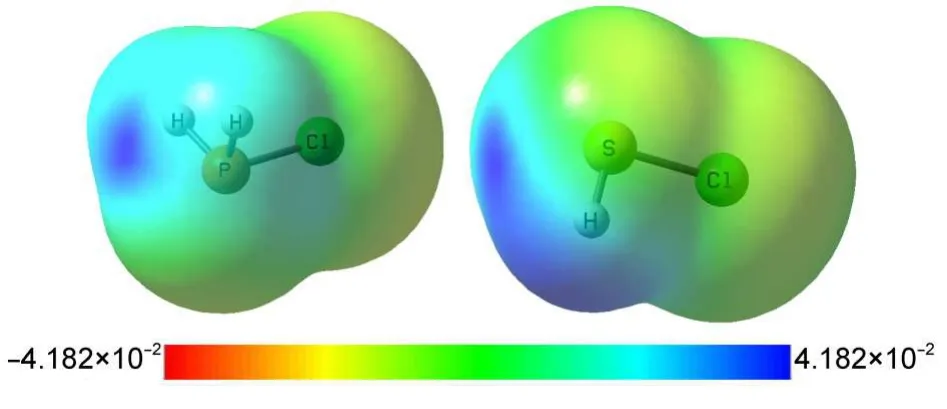
图2 PH2Cl(左)和SHCl(右)单体分子在0.0004 a.u.等电子密度面上分子静电势分布Fig.2 Molecular electrostatic potential(MEP) distribution of PH2Cl(left)and SHCl(right)on 0.0004 a.u.isosurface
3.2 几何结构、频率与相互作用能
表1列出了用MP2/aug-cc-pVDZ方法计算的各类磷键与硫键结构的主要结构参数(单体间P…S距离,磷键或硫键作用角度∠XPS或∠YSP)、键指数(PS、XP与YS的Wiberg键级(WBI))以及复合物形成时PX或SY键长与振动频率的变化.表1也列出了用MP2/aug-cc-pVDZ、MP2/aug-cc-pVTZ和CCSD (T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ三种方法计算的经过BSSE矫正的相互作用能,分别记为E1、E2和E3.另外也用完全基组方法CBS-QB3计算了单体与复合物的能量,用此方法计算的相互作用能记为E4,也列于表1中.关于单体与复合物更加详细的结构数据列在Supporting Information表S1中.不同方法计算的相互作用能有一定的差别,其中完全基组方法CBS-QB3计算的作用能最小,MP2/aug-ccpVTZ计算的作用能最大,其他两种方法的作用能比较接近.由于没有获得相应的实验数据,很难判断用哪种方法计算能量更好.
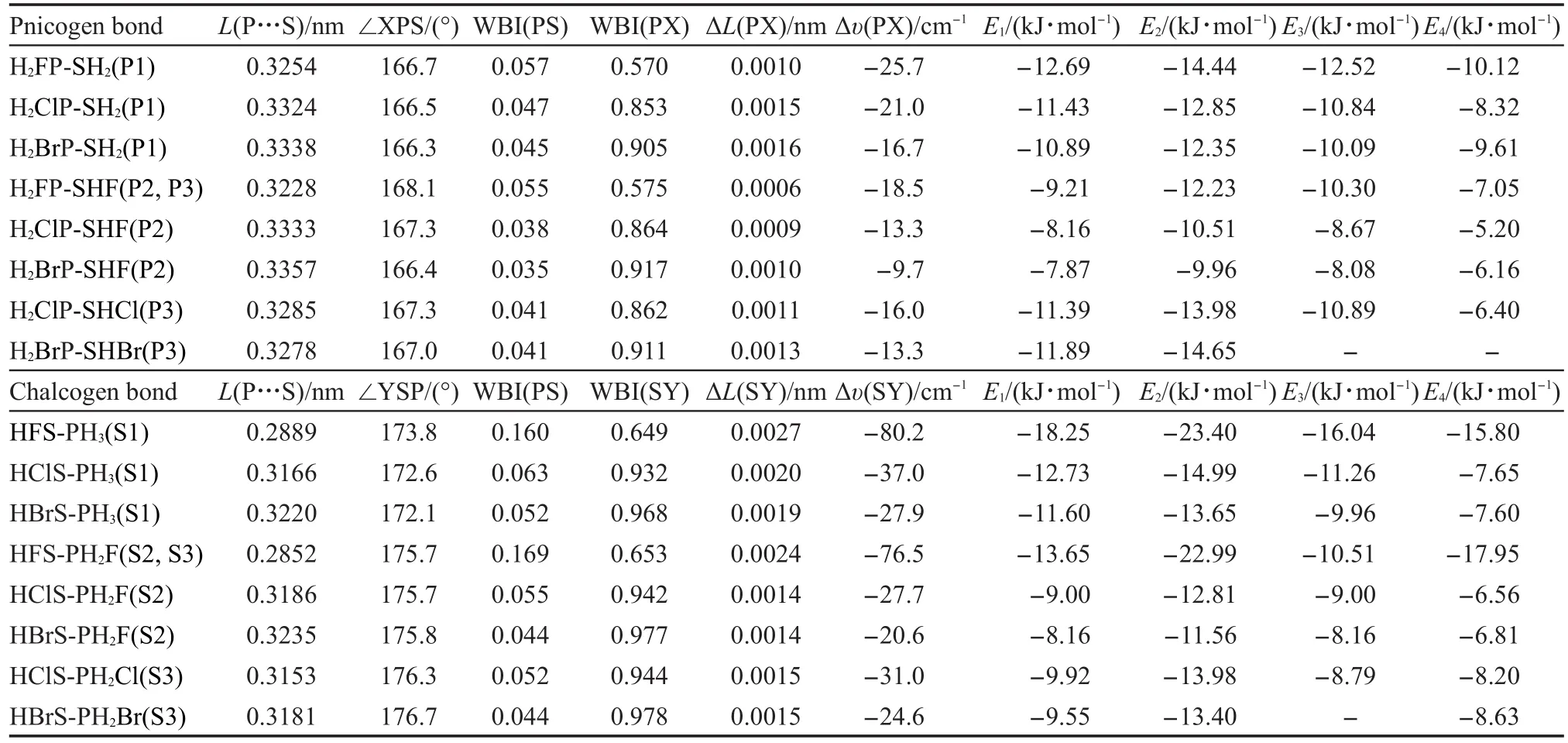
表1 用MP2/aug-cc-pVDZ方法计算的各种H2XP…SHY复合物的主要结构参数和性质与四种方法计算的相互作用能Table 1 Main structural parameters of H2XP…SHY complexes at MP2/aug-cc-pVDZ level and interaction energies calculated using four methods
在磷键体系中磷键的形成导致XP键伸长以及振动频率红移,在硫键体系中硫键的形成导致YS键伸长及其振动频率红移,这分别是由于分子间超共轭n(S)➝σ*(PX)与n(P)➝σ*(SY)产生的电子密度从一个单体的孤对轨道转移到另一个单体的相关反键σ*轨道导致PX或SY键削弱造成的.硫键体系的键长增大与频率红移大于磷键体系,硫键体系中P…S距离比磷键体系的总体来说要小,硫键的键能总体上比磷键大(对称体系P3与S3例外),说明硫键总体上比磷键强.硫键作用角度∠YSP大于磷键的∠XPS,硫键更加接近于直线型,这个差别可能与P和S的价态与配位数不同有关.
对P1、P2、S1与S2型结构,随着取代基X或Y按照F、Cl与Br的顺序变化,P…S距离依次递增,键指数WBI(PS)依次递减,WBI(PX)与WBI(SY)依次递增,SY键的伸长与振动频率红移依次递减,而PX键的伸长依次递增但振动频率红移依次递减,相互作用能依次减小.这些(特别是相互作用能、P…S距离以及WBI等数据)说明形成的磷键或硫键按此顺序越来越弱,磷键或硫键的形成对PX或SY键的削弱越来越弱以及对单体结构和性质的影响也越来越弱,27这是σ-hole的强度按此顺序递减的结果.P2/ S2系列结构形成的磷键/硫键比P1/S1系列对应结构的磷键/硫键弱,这是Lewis碱上电负性大的取代基F吸电子能力强导致Lewis碱的亲核性减弱的结果.
对P3和S3型对称取代结构H2XP…SXH,大电负性的X原子大的吸电子能力使得P(或S)有强的σhole,但也使得S(或P)的亲核能力减弱,这两个作用彼此抵消,最终磷键与硫键的强弱决定于这两个作用的竞争.从表1计算结果看,对硫键S3结构系列,第一个作用整体上占优势,随着取代基X按照F、Cl与Br的顺序变化,P…S距离依次递增,WBI(PS)依次递减,WBI(SY)依次递增,SY键的伸长与振动频率红移依次递减,硫键键能依次递减.而对磷键P3结构系列,这两个作用的表现比较复杂,任一个都没有明显优势,随着取代基X按照F、Cl与Br的顺序变化,WBI(PX)依次递增,PX键伸长依次递增而振动频率红移依次下降,相互作用能依次递增.
此外,在所有各系列的F、Cl与Br取代的三个结构的各种性质中,Cl与Br取代结构的数值比较接近,而F取代结构的相应数值与之差距较大,这是Cl和Br电负性接近而F比它们的电负性大得多的结果.
3.3 磷键与硫键的本质特征
在MP2/aug-cc-pVDZ水平下对所有单体与复合物进行了电子密度拓扑分析与自然间轨道分析,电子密度临界点的性质列于表2中.对于所有的磷键和硫键系列,单体间S…P临界点电子密度ρ<0.1a.u.而拉普拉斯∇2ρ在0-0.1 a.u.之间,因此磷键和硫键都是弱的相互作用,但硫键的电子密度大于磷键的电子密度,说明硫键更强,与前面的分析一致.所有磷键的S…P临界点的电子能量密度H都是一个很小的正值(一个例外),而所有硫键的临界点的电子能量密度H则略大些,是一个小的负值(一个例外).因此大致可以说硫键有很小的共价特征.取代基在硫键系列S1、S2与S3的效应有明显的规律,即从F到Cl和Br,S…P临界点电子密度与拉普拉斯都减小,表明硫键减弱,与前面的结果一致.而对于磷键系列,则取代基的效应没有明显的规律.

表2 用MP2/aug-cc-pVTZ计算的复合物中P…S与PX和SY键临界点的性质Table 2 Characteristics of P…S,PX,and SY bond critical points calculated at MP2/aug-cc-pVDZ level

表3 在MP2/aug-cc-pVDZ水平下计算的LMOEDA能量分解分析结果Table 3 Energy decomposition analysis by LMOEDAcalculated at MP2/aug-cc-pVDZ level
磷键或硫键的形成对单体中化学键PX与SY的影响有一定的规律性.对磷键系列,分子间磷键的形成会导致PX键临界点电子密度降低,且该降低的趋势会随着电子供体SH2分子内的氢原子被卤族原子X(F、Cl、Br)的取代而有所削弱.分子间硫键的形成也具有同样的影响规律,且SY键临界点电子密度下降的幅度更大(只有HFS…PH2F构型中ρ(SY)比HFS…PH3中稍低).
自然键轨道分析表明,在磷键复合物中,有电子密度从S孤对轨道lp(S)向σ*(PX)反键轨道转移,导致lp(S)占据减小而σ*(PX)占据增大,从而PX键伸长并且振动频率红移,分子间超共轭的二阶稳定化能E(2){lp(S)-σ*(PX)}约为31.4-44.0 kJ·mol-1,分子间电荷转移大致为0.03 a.u.左右.在硫键复合物中,存在电子密度从P孤对轨道lp(P)向σ*(SY)反键轨道转移,导致lp(P)占据减小而σ*(SY)占据增大,因而SY键长伸长并且振动频率红移,分子间超共轭的二阶稳定化能E(2){lp(P)-σ*(SY)}较大,约为41.9-132.3 kJ·mol-1,分子间电荷转移也较大(0.04-0.22 a.u.),这些数据也表明硫键比磷键强.
为进一步考察磷键与硫键的本质,我们在MP2/ aug-cc-pVDZ水平下用定域分子轨道能量分解分析方法计算了所有磷键与硫键结构的相互作用能的各个成分(没有进行BSSE校正),结果列于表3中.排斥能(REP)总是绝对值最大的项.在静电能(ES)、交换能(EX)、极化能(POL)与色散能(DISP)四个吸引项中交换项EX贡献最大,达到总吸引能的50%,其次是静电项,贡献为20%-26%,第三是DISP色散项贡献为13%-19%,最小是POL极化项贡献为10%-12%.例外的是两个含F的硫键结构HFS…PH3与HFS…PH2F,其中极化能占总吸引能的15%-16%,而DISP色散能的贡献仅为8%-9%.这些结果与磷键和硫键的一般特征吻合,LMOEDA分析给出的磷键和硫键的一般规律是:排斥项绝对值最大,交换项对总吸引能的贡献最大,一般达到50%,其次是静电项,贡献一般大于20%.
4 结论
从理论上研究了三个磷键与三个硫键系列H2XP…SHY(X,Y=H,F,Cl,Br)的结构、键能、频率与成键特征,发现P或S上的取代基对磷键和硫键的影响具有良好的规律性.随着取代基X(或Y)的电负性增大,P(或S)的σ-hole的亲电性增强,形成的磷键XP…S(或硫键YS…P)逐渐增强,键能逐渐增大,单体间距离P…S减小,PX(或SY)键的伸缩振动频率红移逐渐增大,磷键(或硫键)对单体结构与性质的影响越来越大.总体来说,硫键比磷键强,硫键作用更接近于线性并且有小的共价特征.对磷键与硫键作用能贡献较大的是交换与静电作用.磷键与硫键都是给体-受体相互作用,这种作用导致两个单体有一定程度的极化.
Supporting Information:Some structural parameters of all monomers and complexes are listed in Table S1.This information is available free of charge via the internet at http://www. whxb.pku.edu.cn.
(1)Buckingham,A.D.;Fowler,P.W.;Hutson,J.M.Chem.Rev. 1988,88,963.doi:10.1021/cr00088a008
(2)Chalasinski,G.;Szczesniak,M.M.Chem.Rev.2000,100, 4227.doi:10.1021/cr990048z
(3)Wormer,P.E.S.;van derAvoird,A.Chem.Rev.2000,100, 4109.doi:10.1021/cr990046e
(4)Jeffrey,G.A.Crystal.Rev.2003,9,135.doi:10.1080/ 08893110310001621754
(5)Gilli,G.;Gilli,P.The Nature of the Hydrogen Bond;Oxford University Press:Oxford,UK,2009;pp 2-51.
(6)Bakhmutov,V.I.Dihydrogen Bond:Principles,Experiments, and Applications;Wiley-Interscience:New York,2008.
(7)Politzer,P.;Murray,J.S.;Clark,T.Phys.Chem.Chem.Phys. 2010,12,7748.doi:10.1039/c004189k
(8)Brammer,L.;Espallargas,G.M.;Libri,S.CrystEngComm 2008,10,1712.doi:10.1039/b812927d
(9)Politzer,P.;Murray,J.S.;Clark,T.Phys.Chem.Chem.Phys. 2013,15,11178.doi:10.1039/c3cp00054k
(10)Scheiner,S.CrystEngComm 2013,15,3119.doi:10.1039/ c2ce26393a
(11)Guan,L.Y.;Mo,Y.R.J.Phys.Chem.A 2014,118,8911.doi: 10.1021/jp500775m
(12)Murray,J.S.;Lane,P.;Clark,T.;Politzer,P.J.Mol.Model. 2007,13,1033.doi:10.1007/s00894-007-0225-4
(13)Clark,T.;Hennemann,M.;Murray,J.S.;Politzer,P.J.Mol. Model.2007,13,291.doi:10.1007/s00894-006-0130-2
(14)Murray,J.S.;Lane,P.;Politzer,P.J.Mol.Model.2009,15, 723.doi:10.1007/s00894-008-0386-9
(15)Scheiner,S.J.Phys.Chem.A 2011,115,11202.doi:10.1021/ jp203964b
(16)Scheiner,S.International Journal of Quantum Chemistry 2013, 113,1609.doi:10.1002/qua.v113.11
(17)Alkorta,I.;Sánchez-Sanz,G.;Elguero,J.;Del Bene,J.E.J. Phys.Chem.A 2013,117,183.doi:10.1021/jp3100816
(18)Del Bene,J.E.;Alkorta,I.;Elguero,J.Theor.Chem.Acc.2014, 133,1464.doi:10.1007/s00214-014-1464-y
(19)Adhikari,U.;Scheiner,S.J.Phys.Chem.A 2014,118,3183.
(20)Boys,S.F.;Bernardi,F.Mol.Phys.1970,19,553.
(21)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, RevisionA.02;Gaussian:Wallingford,CT,2009.
(22)Keith,T.A.AIMAll,Version 13.10.19;TK Gristmill Software, Overland Park KS,USA,2013.(aim.tkgristmill.com).
(23)Glendening,E.D.;Badenhoop,J.K.;Reed,A.E.;Carpenter,J. E.;Bohmann,J.A.;Weinhold,F.GENNBO5.0W;Theoretical Chemistry Institute:University of Wisconsin,Madison,WI, 2001.
(24)Su,P.;Li,H.J.Chem.Phys.2009,131,014102.doi:10.1063/ 1.3159673
(25)Schmidt,M.W.;Baldridge,K.K.;Boatz,J.A.J.Comput. Chem.1993,14,1347.
(26)Lu,T.;Chen,F.J.J.Comput.Chem.2012,33,580.doi: 10.1002/jcc.v33.5
(27)Zhao,G.;Han,K.Accounts Chem.Res.2012,45,404.doi: 10.1021/ar200135h
Theoretical Analysis of Pnicogen and Chalcogen Bonds in H2XP…SHY Complexes
LIU Yu-Zhen LI An-Yong*
(School of Chemistry and Chemical Engineering,Southwest University,Chongqing 400715,P.R.China)
The MP2 and CCSD(T)ab initio quantum chemistry methods were applied to study the pnicogen bonds X—P…S and chalcogen bonds Y—S…P formed between PH2X and SHY(X,Y=H,F,Cl,Br)and the effects of the substituents X and Y on the bonds.Calculated results show that the chalcogen bonds are stronger than the pnicogen bonds.Strongly electronegative substituents that are connected to the Lewis acid strengthened the bonds and significantly affected the structures and properties of the monomers.Conversely, the substituents connected to the Lewis bases produced opposite effects.The energies of chalcogen bonds were 8.37-23.45 kJ·mol-1;the strongest chalcogen bond was found in the structure HFS-PH3using the CCSD (T)method with a bonding energy of 16.04 kJ·mol-1.The energies of pnicogen bonds were in the range 7.54-14.65 kJ·mol-1;the strongest pnicogen bond was found in H2FP-SH2using CCSD(T)with a bonding energy 12.52 kJ·mol-1.The most important factors for bond strength for both types of bonds were the exchange and electrostatic energies.The hyperconjugations lp(S)-σ*(PX)and lp(P)-σ*(SY)play important roles in the formation of the pnicogen and chalcogen bonds,which both lead to polarization of the monomers.Polarization caused by the chalcogen bond is larger than that by the pnicogen bond,resulting in the chalcogen bond having less of a covalent character.
σ-hole;Pnicogen bond;Chalcogen bond;Substituent effect
O641
10.3866/PKU.WHXB201501211www.whxb.pku.edu.cn
Received:December 1,2014;Revised:January 21,2015;Published on Web:January 21,2015.
∗Corresponding author.Email:aylifnsy@swu.edu.cn;Tel:+86-23-68252360
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
药学进展(2022年1期)2022-03-06
潍坊学院学报(2021年2期)2021-07-22
国防科技大学学报(2020年6期)2020-12-07
山西卫生健康职业学院学报(2020年6期)2020-04-27
硅酸盐通报(2020年1期)2020-02-25
测绘通报(2019年11期)2019-12-03
赤峰学院学报·自然科学版(2019年5期)2019-09-10
食品科学(2019年14期)2019-07-26
