
靶向特定氨基酸的共价抑制剂研究进展
2022-03-06 09:11王傲雪裴俊平王贯欧阳亮
药学进展 2022年1期
王傲雪,裴俊平,王贯,欧阳亮
(四川大学华西医院生物治疗国家重点实验室,四川 成都610041)
共价抑制剂是通过共价键与靶蛋白氨基酸残基产生更牢固的结合作用,从而更好地发挥靶蛋白抑制活性的一类小分子化合物。传统的抑制剂通过氢键与范德华力等非共价相互作用实现对靶蛋白的结合与抑制,较小的作用强度往往使得此类药物具有较低的选择性与抑制能力。考虑到共价键的强度远高于非共价相互作用,共价抑制剂显然具有更强的抑制潜力。并且与非共价抑制剂相比,共价结合的药物不符合标准平衡动力学,药物被排出体外后仍能保持抑制效果,这意味着系统的药物暴露和非靶药物结合减少,从而使得毒性降低[1]。然而,共价抑制剂的脱靶效应以及对蛋白质的非特异性修饰等现象仍然是研究人员担忧的问题,因此,长期以来很少有药物被有意设计为共价抑制剂。
尽管如此,仍有许多小分子共价抑制剂上市。最早的共价抑制剂可以追溯到阿司匹林。早在19世纪初,其已经被用来治疗疼痛和发热,而直到20世纪70年代经研究才发现阿司匹林以与丝氨酸残基共价结合的方式不可逆抑制乙酰化环氧合酶[2]。英国医生于20世纪偶然发现的抗生素青霉素也是共价抑制剂,其通过β-内酰胺环不可逆地与青霉素结合蛋白的丝氨酸残基形成共价键,阻止细胞壁合成[3]。阿法替尼被美国FDA批准用于治疗相应转移性非小细胞肺癌,其能够不可逆地与表皮生长因子受体(epidermal growth factor receptor,EGFR)的半胱氨酸形成共价键[4]。硼替佐米通过硼化作用可逆地与26S蛋白酶体活性位点的β5与β5i亚基中的苏氨酸共价结合,并于2003年被美国FDA批准用于治疗复发或难治性骨髓瘤[5]。这些药物都是医药界不可或缺的重要组成部分。对共价抑制剂的研究,证明了其具有降低给药浓度、延长有效治疗时间、减少耐药性、增加不可成药靶点的开发可能性等明确优势[6-8]。随着人们对共价抑制剂机制的研究以及对亲电弹头与亲核氨基酸的探索,越来越多的共价抑制剂得到开发。根据文献调研,已有至少50种共价抑制剂获批上市用于治疗各类疾病[9]。本文将重点介绍已经被美国FDA批准上市或进入临床的共价抑制剂以及具有临床应用潜力的共价抑制剂的研究近况,以期为新型共价抑制剂的开发提供依据与线索。
1 共价抑制剂的作用机制
共价抑制剂通常具有导引头和弹头2个结构,其通过2个相关却不连续的步骤完成与靶蛋白的共价结合:1)小分子配体会通过导引头与靶蛋白特定的结合口袋可逆地发生非共价相互作用,形成中间体E·I;2)中间体复合物中小分子配体的亲电弹头会与靶蛋白的亲核氨基酸残基通过加成、取代、氧化等反应发生共价相互作用,生成共价复合物E-I,这一步反应通常比第1步慢。根据第2步反应是否可逆,可将共价抑制剂分为不可逆共价抑制剂和可逆共价抑制剂,其作用模式如图1所示。在第2步中,K2,K-2分别代表生成速率常数与解离速率常数。当K-2不等于0时,该化合物为可逆共价抑制剂;当K-2等于或趋近于0时,该化合物为不可逆共价抑制剂[1,10-12]。
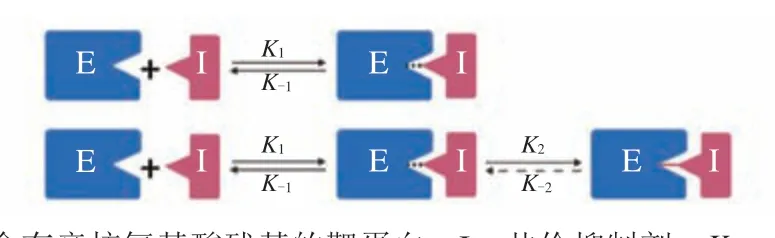
图1 共价抑制剂与靶蛋白相互作用机制Figure 1 Mechanism of the interaction between covalent inhibitor and target protein
不可逆共价抑制剂包含的亲电弹头有丙烯酰胺、α-卤酮、环氧化物、氮丙环、乙烯基砜、活化乙炔等,大多用于激酶类抑制剂的设计。只有在机体形成新的蛋白或原蛋白复合物降解时才能解除此类化合物的抑制作用,这也使得不可逆共价抑制剂能够产生持久又强大的效力,从而降低服用剂量与次数,提高患者的依从性。
可逆共价抑制剂包含的亲电弹头通常有醛类、活化酮类、α-酮基酰胺类、腈类和硼酸衍生物等,多用于蛋白酶体、丝氨酸水解酶、组织蛋白酶等靶标的可逆共价抑制剂的设计。这类抑制剂与靶标产生可逆的共价结合,药动学性质在不可逆抑制剂与非共价抑制剂之间,使其不仅兼备了不可逆抑制剂的优点,也减少了因脱靶效应导致的潜在风险。常见靶向特定氨基酸的共价基团分类如表1所示。

表1 靶向特定氨基酸的共价基团分类Table 1 Classification of covalent groups targeting specific amino acids
2 靶向亲核基团含硫元素氨基酸的共价抑制剂
2.1 靶向半胱氨酸的共价抑制剂
半胱氨酸(cysteine,Cys)的硫醇基团亲核性并不高,而当其侧链中的硫醇基团以巯基形式存在时,亲核能力会大大提升,并使其一跃成为20种常见氨基酸亲核性之首,这也使得半胱氨酸成为最为普遍靶向的共价氨基酸残基[13-14]。强亲核能力意味着弱亲电弹头的小分子同样能够作为共价抑制剂被开发,降低了脱靶的风险。目前靶向半胱氨酸的抑制剂有EGFR、布鲁顿酪氨酸激酶(Brutons tyrosine kinase,BTK)、成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)、Kirsten肉瘤病毒致癌基因同源物(Kirsten rat sarcoma viral oncogene homolog,KRAS)、丝裂原活化蛋白激酶1(mitogen-activated protein kinase 1,MAPK1)、核输出蛋白1(exportin 1,XPO1)、p90核糖体S6激酶(p90 ribosomal S6 kinase,RKS)、含NLR家族Pyrin域蛋白3(NLR family pyrin domain containing protein 3,NLRP3)、中心 体相 关激 酶2(NIMA related kinase 2,NEK2)、Janus激酶3(Janus kinase 3,JAK3)抑制剂等[11,15-22],本文将选取其中部分经典共价抑制剂进行介绍。
2.1.1 表皮生长因子受体共价抑制剂EGFR是多种癌症的关键治疗靶点,研究人员致力于开发 EGFR抑制剂或双靶抑制剂进行相关疾病的治疗[23-24]。但其激酶结构域经常发生突变而对多种EGFR抑制剂产生耐药。共价EGFR抑制剂能够很好地降低耐药可能性,且目前已有许多策略被用于减少其脱靶的风险。
2.1.1.1 不可逆共价抑制剂这类EGFR共价抑制剂通常将迈克尔受体基团作为亲电弹头,其中心杂环支架的N原子能够与EGFR形成氢键,增加小分子的停留时间。阿法替尼(afatinib,1)是首个上市的第2代EGFR共价抑制剂,被FDA批准用于治疗EGFR 19外显子缺失或21外显子(L858R)突变的转移性非小细胞肺癌[4]。阿法替尼的迈克尔受体基团为丙烯酰胺基团,其与靶蛋白结构中的氨基酸残基Cys797通过1,4共轭加成反应形成共价键(见图2)。临床前数据表明,阿法替尼对EGFRL858R/T790M突变体激酶的IC50为10 nmol · L-1,约为非共价抑制剂吉非替尼抑制效力的100倍,并在EGFRL858R/T790M突变体以及19外显子缺失的小鼠模型中能显著抑制肿瘤生长[15]。这主要归因于共价抑制剂效率的提高和共价复合物形成的持久性,因为突变能够增强EGFR对腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)的亲和力而导致非共价抑制剂耐药,而共价键的形成能够降低EGFR对ATP浓度的敏感性。作为第3代EGFR抑制剂,奥希替尼(osimertinib,2)于2015年被美国FDA批准治疗转移性EGFRT790M突变阳性的非小细胞肺癌[25]。研究表明,奥希替尼对L858R/T790M EGFR突变体激酶的IC50为1 nmol · L-1,抑制效力约为野生型的200倍。奥希替尼显著下调多种肿瘤细胞中的EGFR磷酸化水平并在相应小鼠模型中抑制肿瘤的生长。其结构中的丙烯酰胺亲电靶头指向Cys797并与之共价结合,以此发挥强效治疗作用[26]。然而临床数据显示,许多EGFRT790M突变的患者会在治疗中产生C797S突变,诱导奥西替尼耐药性的产生[27]。

图2 化合物1与表皮生长因子受体的共晶结构(PDB ID:4G5P)Figure 2 Co-crystal structure of compound 1 with EGFR (PDB ID: 4G5P)
2.1.1.2 可逆共价抑制目前仍未有可逆EGFR共价抑制剂上市,但已有许多抑制剂对突变EGFR具有高度选择性及良好的抑制能力,如化合物3,其能够与EGFR中的氨基酸残基Cys797共价结合,并具有更高的选择性,对EGFRL858R的IC50为0.15 μmol · L-1,对EGFRL858R/T790M的IC50为0.037 μmol · L-1[25,28]。
2.1.2 布鲁顿酪氨酸激酶共价抑制剂BTK能够影响B淋巴细胞的发育和功能,是血液系统恶性肿瘤的关键靶点,并能够导致自身免疫性疾病的发生。近年来科研人员已经开发了大量小分子BTK共价抑制剂以及蛋白降解靶向嵌合体(proteolysis-targeting chimera,PROTAC),为相关疾病的治疗提供了新的策略[16,29-32]。
2.1.2.1 不可逆共价抑制剂2013年,伊布替尼(ibrutinib,4)被批准治疗B淋巴细胞恶性肿瘤。其对BTK具有较高选择性,并对BTK有着较高的抑制效力(IC50= 0.5 nmol · L-1),临床前数据表明其在自身免疫性疾病和B淋巴细胞恶性肿瘤动物模型中十分有效[29]。伊布替尼中的丙烯酰胺弹头与BTK蛋白ATP结合域中的氨基酸残基Cys481发生共价结合,发挥对BTK的抑制作用,然而该抑制剂能够以时间依赖的方式与非靶标蛋白发生反应。并且由于不能与丝氨酸的羟基形成共价键,伊布替尼对BTK C481S突变的抑制效力大大下降(IC50= 1 μmol · L-1)。作为第2代BTK抑制剂,阿卡替尼(acalabrutinib,5)于2017年被美国FDA批准上市,其对BTK具有更高的选择性、更强的抑制能力和更小的副作用。其同样靶向BTK的Cys481,对BTK的IC50为3 nmol · L-1,在人全血CD69 B淋巴细胞活化试验中,其EC50为8 nmol · L-1[30,33]。
2.1.2.2 可逆共价抑制剂由于伊布替尼带来的非靶效应相关的毒性作用,FDA并未批准其治疗自身免疫性疾病,因此,研究人员深入研究了BTK可逆共价抑制剂,以期为相关疾病的治疗做出贡献。2017年,PRN1008(6)用于健康志愿者的Ⅰ期临床试验表明,该药具有较高的安全性与良好的耐受性[34]。研究结果显示,PRN1008在外周血单核细胞中缓慢持续结合BTK蛋白,而不形成永久性共价键。PRN1008对BTK有较高抑制能力(IC50= 1.3 nmol · L-1),对人B淋巴细胞瘤细胞的IC50为7.4 nmol · L-1[35]。PRN1008在临床前药效学研究中表现出逆时针滞后的现象,能够减少药物的长期体循环,从而降低潜在的脱靶副作用。

2.1.3 KRAS共价抑制剂KRAS是原癌基因RAS中突变频率最高的亚型,多年来一直被认为是一个不可成药的靶标。KRASG12C是最常见的一种突变体,半胱氨酸的存在使得突变KRAS的选择性共价靶向成为可能[17,36-38]。
2018年,美国安进公司通过基于具有半胱氨酸反应功能的片段筛选与结构设计开发得到高效、高选择性、耐受性良好的共价抑制剂AMG510(7),这是FDA批准的首个KRASG12C不可逆共价抑制剂。临床前研究表明,AMG510对MIA PaCa-2细胞中的细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)的IC50为68 nmol · L-1,对MIA PaCa-2细胞也具有显著的抑制增殖作用,IC50为5 nmol · L-1。在KRASG12C突变肿瘤模型中,发现其能够持续抑制MAPK信号通路。当经口给药剂量大于10 mg · kg-1时,观察到小鼠体内ERK磷酸化被显著抑制。在长达24 d的治疗时间里,AMG510显著持续抑制小鼠肿瘤的生长。尽管AMG510半衰期相对较短,但由于KRAS蛋白半衰期长达22 h,AMG510的快速共价灭活也能够产生持久的药学效应[39]。除此之外,威尔斯普林公司与米拉提治疗公司开发的ARS-1620(8)及MRTX849(9)同样也进入Ⅰ期临床试验[38,40]。礼来公司研发的LY3537982在临床前研究中显示出极佳的抗肿瘤活性,对具有KRASG12C突变的H23肺癌细胞系展现出较高的抑制效力(IC50= 1.04 nmol · L-1),并能诱导异种移植模型中肿瘤的完全消退。

2.1.4 其他靶点的共价抑制剂XPO1对维持细胞内稳态十分重要,在许多肿瘤细胞中都观察到XPO1的过表达,并与包括血液恶性肿瘤在内的多种肿瘤的发生发展相关联。目前已经有许多选择性XPO1抑制剂得到开发,但仅有抑制剂selinexor(10)通过临床试验,并于2019年被美国FDA批准与地塞米松联用治疗患有复发或难治性多发性骨髓瘤的成年患者[41]。Selinexor能够选择性靶向XPO1,并与XPO1的氨基酸残基Cys528发生缓慢可逆的共价结合[42]。目前针对骨质疏松已有许多药物上市, 组织蛋白酶K(Cathepsin K,Cat K)是治疗骨质疏松的关键靶点,其可逆性共价抑制剂奥当卡替(odanacatib,11)正在进行临床研究,用以减少老年妇女的骨折发生[14,43]。其结构中的腈基与Cat K活性口袋中半胱氨酸的共价结合是可逆的,这可能降低其在人体内的潜在副作用。富马酸二甲酯(12)早已被应用于治疗银屑病和多发性硬化病。2018年,Andersen等[18]发现其能够作为迈克尔受体与核糖体S6激酶2(ribosomal S6 kinase 2,RSK2)中的Cys599共价结合,这使得其能够高效抑制RSK/有丝分裂原和应激活化型蛋白激酶(mitogen-and stress-activated protein kinase,MSK)。

2.2 靶向甲硫氨酸的共价抑制剂
甲硫氨酸(methionine,Met)在脊椎动物中十分稀有,其同样含有硫原子,但其硫醚基团仅具有中等亲核能力,且甲硫氨酸因其疏水性通常并不存在于蛋白表面,这也使得靶向甲硫氨酸的共价抑制剂开发并不常见。
溴结构域蛋白4(bromodomain-containing protein 4,BRD4)是一种关键的表观遗传调控因子,能够影响细胞增殖、迁移与周期等,这也使得其表达失调与多种肿瘤的发生发展密切相关。2018年,Kharenko等[44]开发出了一系列选择性BRD4共价抑制剂,如化合物13,其能够选择性地与BRD4的Met149形成不可逆共价键(见图3),其IC50为0.05 μmol · L-1,并在MV4-11细胞中表现出比可逆抑制剂更加持久的抗肿瘤细胞增殖的能力。

图3 化合物13与溴结构域蛋白4的共晶结构(PDB ID:6CZU)Figure 3 Co-crystal structure of compound 13 with BRD4 (PDB ID: 6CZU)
3 靶向亲核基团含氮元素氨基酸的共价抑制剂
3.1 靶向赖氨酸的共价抑制剂
与半胱氨酸相比,赖氨酸(lysine,Lys)的丰度更高,其残基中的伯胺带有潜在亲核性,能够与一些带有亲电弹头的小分子抑制剂发生不可逆共价结合作用。然而,在生理条件下,氨基往往以质子化形式存在,这为靶向赖氨酸的共价抑制剂开发带来了挑战。为了成功靶向赖氨酸残基,其解离常数需产生变化从而使其在生理环境下保持中性,这意味着靶蛋白的赖氨酸残基需在生理环境中被干扰,或与配体结合时被干扰[13]。
3.1.1 磷酯酶A2共价抑制剂首个靶向赖氨酸残基的共价抑制剂为1977年从帕劳的变异丝瓜菌中分离得到的天然二倍半类萜烯抗生素曼诺力得(manoalide,14)。其被认为能够与磷酯酶A2(phospholipase A2,PLA2)中的氨基酸残基Lys6和Lys79发生不可逆共价结合,并在体外表现出对金黄色葡萄球菌显著的增殖抑制作用,IC50为2 ~ 4 nmol · L-1[45]。
3.1.2 磷脂酰肌醇-3激酶共价抑制剂1957年首次分离得到的甾体类天然产物渥曼青霉素(wortmannin,15)是磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)的高效不可逆抑制剂[46]。化合物15通过氮杂迈克尔加成与配体呋喃环的开环对PI3K活性位点的Lys833进行共价修饰(见图4),对PI3K的IC50为2 ~ 4 nmol · L-1。但是其对PI3K具有较低的选择性,并且稳定性不高。为了克服这些问题,进一步开发得到其开环类似物,其能够通过乙烯基酰胺转移反应与赖氨酸位点不可逆共价结合,并已经进入非小细胞癌、去势抵抗性前列腺癌与胶质母细胞瘤的临床试验[47]。2018年,Dalton等[48]开发了一系列选择性不可逆PI3Kδ共价抑制剂,如化合物16,其对PI3Kδ的pIC50为4.8。研究表明其能够与PI3Kδ亚型的Lys779反应,并在浓度低于1 μmol · L-1时对活细胞中的PI3Kδ具有高度选择性。同年,提取自沙生盐藻CNY-486的化合物neolymphostin A(17)作为PI3K和雷帕霉素受体蛋白(mammalian target of rapamycin,mTOR)双重选择性共价抑制剂得到开发,其同样也是首个来自细菌的共价激酶抑制剂[49]。Neolymphostin A能够通过结构中的亲电乙烯基酯共价靶向Lys802,并以3 nmol · L-1的浓度阻断活细胞中的蛋白激酶B(protein kinase B,AKT)磷酸化。

图4 化合物15与磷脂酰肌醇-3激酶的共晶结构(PDB ID:1E7U)Figure 4 Co-crystal structure of compound 15 with PI3K (PDB ID: 1E7U)
3.1.3 真核翻译起始因子4E共价抑制剂真核翻译起始因子4E(eukaryotic translation initiation factor 4E,elF4E)能够调节依赖性蛋白质翻译的起始阶段,研究表明,elF4E在多种肿瘤中表达水平升高,并与肿瘤的恶性进展有关。2020年,Wan等[50]为了克服现有elF4E抑制剂细胞渗透性低等问题,利用共价对接的方法开发了首个具有细胞活性的elF4E共价抑制剂。化合物18能够与elF4E中的Lys162发生共价结合。尽管其稳定性不高,但对赖氨酸残基的高亲和力能够使其尽快完成共价修饰。
3.1.4 X连锁凋亡抑制蛋白共价抑制剂2018年,Baggio等[51]利用热力学方法开发了首个有效靶向赖氨酸的共价X连锁凋亡抑制蛋白(X-linked inhibitor-of-apoptosis protein,XIAP)抑 制 剂。其中,化合物19能够与杆状病毒IAP重复序列3(baculovirus IAP repeat 3,BIR3)结 构 域 中 的Lys311共价结合,并对其产生显著的抑制作用(IC50= 16.6 nmol · L-1)。在对LCL161耐药的L363细胞中,化合物19显示出极强的肿瘤细胞抑制能力,在浓度为20 μmol · L-1条件下处理48 h能够抑制70%以上的细胞的生长。在研究中发现化合物19与BIR3结构域的共价结合过程十分缓慢,为了克服这一问题,Baggio等[52]于2019年开发了新的具有细胞抑制效力的芳基氟硫酸盐类共价抑制剂化合物20。化合物20能够与BIR3结构域中的Lys297残基毗邻,这也使得其在激酶以及细胞层面更容易发生共价反应。化合物20在6 h处理条件下对XIAP的IC50为12 nmol · L-1,在水性缓冲溶液中溶解度为2 mmol · L-1,并在血浆中能保持稳定[半衰期(t1/2)>2 h]。在化合物21与肿瘤坏死因子α同时存在的条件下,能够高度诱导SK-MEL-28细胞系与A549细胞系的凋亡,这表明化合物20与临床候选药物LCL161具有相当的细胞药理学特性。
3.1.5 骨髓细胞白血病蛋白1共价抑制剂2016年,Akcay等[53]首次开发了靶向骨髓细胞白血病蛋白1(myeloid cell leukemia 1,Mcl-1)的可逆共价抑制剂,其能够通过芳基硼酸羰基弹头共价修饰Lys234,并显示出比非共价同源物更强的抑制能力。化合物21对Mcl-1的IC50为3.4 nmol · L-1。其通过对Mcl-1的抑制作用在MOLP-8细胞中激活半胱天冬酶-3/7来促进细胞凋亡(EC50= 0.46 μmol · L-1),这比其他非共价抑制剂活性提高了24倍以上。2021年,Gambini等[54]开发了首个共价靶向Mcl-1的Lys残基的BH3肽,该肽对hMcl-1具有显著的亲和力和选择性。
3.2 靶向组氨酸的共价抑制剂
组氨酸(histidine,His)中的咪唑基团带有亲核性,其能够与磺酰氟、螺环氧化物、α,β-不饱和酮或醛等发生反应形成共价键。研究证明,相比于其他亲核试剂,组氨酸残基能够与亲电性相对较弱的试剂特异性结合,使得这些试剂具有被开发为共价抑制剂的可能性。尽管如此,目前对于靶向His氨基酸残基的共价抑制剂的开发并不多。
3.2.1 甲硫氨酸氨基肽酶2共价抑制剂从烟曲霉中提取得到的天然产物烟曲霉素的类似物beloranib(22)是甲硫氨酸氨基肽酶2(methionine aminopeptidase 2,MetAP2)的共价抑制剂,与其结构中阻碍较小的螺环氧化合物能够与MetAP2活性位点上的His231产生共价结合,尽管化合物22在治疗癌症方面效果不明显,但在研究中发现其具有降低体质量的作用,并且耐受性良好。目前22已经作为肥胖症治疗的候选药物进入Ⅲ期临床研究,在临床试验中没有发现与治疗相关的明显副作用[55-57]。

4 靶向亲核基团含碳和氧元素的氨基酸共价抑制剂
4.1 靶向丝氨酸的共价抑制剂
丝氨酸(serine,Ser)是蛋白酶和其他水解酶活性位点的关键催化残基,其侧链上的羟基能够被邻近残基激活,从而和带有亲电弹头的小分子产生共价结合。
4.1.1 青霉素结合蛋白共价抑制剂1928年发现的青霉素(penicillin,23)作为抗生素早已应用于临床治疗多年。研究发现化合物23的β-内酰胺环能够通过酰化作用共价靶向青霉素结合蛋白(penicillinbinding protein,PBP)活性位点Ser36,通过这种方式青霉素能够抑制细菌合成细胞壁从而发挥对应的抗菌作用[3]。
4.1.2 β-内酰胺酶共价抑制剂β-内酰胺类抗生素在治疗过程中出现了耐药现象,这是由于细菌能够通过表达β-内酰胺酶使得这类抗生素中的β-内酰胺水解从而失去对细菌的抑制能力。为了克服这种耐药性,靶向β-内酰胺酶的抑制剂应运而生。阿维巴坦(avibactam,24)是一种可逆共价抑制剂,其五元环脲中的羰基能够与靶蛋白活性部位的Ser残基产生共价相互作用并使其乙酰化[58]。阿维巴坦与头孢噻林的联用在针对革兰阴性菌感染患者的Ⅱ期临床试验中表现出良好的疗效,并于2015年被美国FDA批准上市。
4.1.3 乙酰胆碱酯酶共价抑制剂阿尔茨海默病仍是难以攻克的神经退行性疾病,除抑制β-淀粉样蛋白聚集外,靶向乙酰胆碱酯酶(acetylcholine esterase,AchE)同样能够对这种疾病进行一定程度的治疗[59]。天然产物硬豆碱的类似物卡巴拉汀(rivastigmine,25)是一种共价靶向AchE的抑制剂,其能够选择性地作用于中枢神经系统中的AchE[60]。卡巴拉汀已经被批准用于阿尔茨海默病的治疗,其能够通过结构中的酚类氨基甲酸酯酰化AchE活性位点的氨基酸残基Ser203,抑制时间可达10 h[61]。

4.1.4 丙型肝炎病毒蛋白酶共价抑制剂丙型肝炎病毒蛋白酶(hepatitis C virus,HCV)能够诱导丙型肝炎病毒的复制以及病毒复合物的形成,通过抑制HCV能够治疗相关的感染。
2006年,Perni等[62]开发了特拉匹韦(telaprevir,26)作为HCV的可逆共价抑制剂,特别是选择性靶向HCV NS3-4A。在临床前研究中,化合物26能够在1b型HCV复制子细胞中表现出显著的抗病毒活性(IC50= 354 nmol · L-1),并在动物模型中显示良好的药动学特性。化合物26结构中的α-酮酰胺弹头通过1,2加成到Sp2原子共价靶向HCV中的Ser1 139,并且已经被批准上市。同年,Venkatraman等[63]通过筛选小分子化合物库得到先导化合物,对其进行结构优化后得到HCV NS3选择性抑制剂波普瑞韦(boceprevir,27)。研究发现其具有较高的口服生物利用度,其结构中的亲电酮酰胺可逆地共价靶向Ser139,对人中性粒细胞弹性蛋白酶(human neutrophil elastase,HNE)/HCV选择性较高,并在复制子分析中显示出较高的细胞效力(EC90= 0.35 μmol · L-1)。波普瑞韦已于2011年被批准上市。那拉匹韦(narlaprevir,28)同样具有酮酰胺结构,其在反应过程中被重排与活性位点的Ser139形成可逆共价键(见图5)。那拉匹韦抑制复制子RNA的EC90为40 nmol · L-1。该药于2016年被FDA批准上市[64]。

图5 化合物28与丙型肝炎病毒NS3-4a蛋白酶的共晶结构(PDB ID: 3LON)Figure 5 Co-crystal structure of compound 28 with HCV NS3-4a protease (PDB ID: 3LON)
4.2 靶向苏氨酸的共价抑制剂
苏氨酸(threonine,Thr)的侧链羟基同样能够被邻近的残基激活从而提高其亲核性,这也使得其能够作为亲核氨基酸被带有亲电弹头的抑制剂共价靶向。
4.2.1 蛋白酶体抑制剂蛋白酶体是在细胞内降解蛋白质的一类复合物,在真核生物、古细菌以及某些放线菌中普遍存在。其能够通过对蛋白质的降解调控包括基因表达与细胞周期在内的多种细胞进程,这也使得蛋白酶体的表达水平与许多疾病的发生发展相互关联。由于其活性位点具有亲核Thr残基,设计共价结合Thr的抑制剂已经成为了靶向蛋白酶体从而治疗癌症等相关疾病的一种策略。
4.2.2 可逆共价抑制剂2003年,硼替佐米(bortezomib,29)作为首个蛋白酶体共价抑制剂被美国FDA批准用于治疗复发或难治性骨髓瘤。作为一种二肽硼酸类化合物,硼替佐米能够通过硼化作用可逆地与26S蛋白酶体活性位点的β5与β5i亚基中的苏氨酸共价结合,通过对蛋白酶体的抑制发挥抗肿瘤活性[65-66]。其通过静脉注射或者皮下注射给药。临床前研究表明其对乳腺癌细胞MCF-7的IC90为0.05 mmol · L-1,对非小细胞肺癌细胞H460的IC50为0.1 μmol · L-1,并展现出对骨髓瘤的良好疗效[5,67]。然而,在长期的治疗过后,许多患者出现了对硼替佐米的耐药性,这可能部分由于编码β5亚基的PSMB5基因产生了突变。2015年,伊沙佐米(ixazomib,30)作为首个口服蛋白酶体抑制剂被美国FDA批准用于至少接受过1次既往治疗的多发性骨髓瘤患者的治疗[68]。伊沙佐米在血浆中能够水解为硼酸,可逆地共价靶向20S蛋白酶体的β5亚基,从而发挥对蛋白酶体的抑制作用。其对20S蛋白酶体的β5亚基的抑制能力较高(IC50= 3.4 nmol · L-1)[69]。在临床前研究中发现其能够抑制核因子κB(nuclear factor kappa-B,NF-κB)通路,并诱导多发性骨髓瘤细胞的凋亡。

4.2.3 不可逆共价抑制剂卡非佐米(carfilzomib,31)于2012年被批准通过静脉注射治疗多发性骨髓瘤。研究表明,作为一种环氧酶素衍生物,卡非佐米通过结构中的环氧酮与β5亚基中的Thr催化残基发生不可逆的共价相互作用(见图6)。在临床前研究中,卡非佐米体现出对硼替佐米耐药细胞更强的抑制活性和更长的抑制时间,并对蛋白酶体具有更高的选择性。其对20S蛋白酶体β5的IC50为3.4 nmol · L-1。在临床试验中,卡非佐米具有良好的疗效以及耐受性[70]。

图6 化合物31与20S蛋白酶体的共晶结构(PDB ID: 4R67)Figure 6 Co-crystal structure of compound 31 with 20S proteasome (PDB ID: 4R67)
4.3 靶向酪氨酸的共价抑制剂
酪氨酸(tyrosine,Tyr)中同样含有羟基,亲核性相对较低。但酪氨酸残基存在于蛋白质中时,容易形成高度亲核性的苯氧基阴离子,使得其能够被多种路易斯酸共价靶向。
4.3.1 SR-蛋白特异性激酶1/2抑制剂Hatcher等[71]于2018年开发了首个共价靶向SR-蛋白特异性激酶1/2(SRSF protein kinase 1/2,SPRK1/2)的不可逆抑制剂SRPKIN-1(32),其对SRPK1的IC50为 11 nmol · L-1。SRPKIN-1能够与SPRK1/2的ATP结合口袋中酪氨酸的酚羟基共价结合,从而发挥对SPRK1/2的抑制作用。研究表明,SRPKIN-1能有效地将血管内皮生长因子(vascular endothelial growth factor,VEGF)从促血管生成亚型VEGFA165a转化为抗血管生成亚型VEGF-A165b,并在年龄相关性黄斑变性小鼠模型中以剂量依赖的方式抑制血管的形成。
4.3.2 B细胞淋巴瘤6共价抑制剂2020年,Teng等[72]通过将磺酰氟引入已有的可逆B细胞淋巴瘤6(B-cell lymphoma 6,BCL6)抑 制 剂,开 发 出BCL6共价抑制剂TMX-2164(33)。其能够通过结构中的磺酰氟与BCL6中的Tyr58的羟基发生共价结合。TMX-2164能有效抑制BCL6的激酶活性(IC50= 251 nmol · L-1),并在DLBCL细胞中显示出较高的抗增殖活性。

4.3.3 Ras相关蛋白共价抑制剂由于Ras相关蛋白(Ras-related protein,Ral)缺乏亲核半胱氨酸残基,Bum-Erdene等[73]利用作用于酪氨酸的亲电弹头芳基磺酰氟共价靶向Ras 样GTP水解酶(Ras-like guanosine triphosphatase,RAL GTPase)的氨基酸残基Tyr82(见图7)。化合物34能够抑制鸟嘌呤交换因子Rgl2介导的Ral GTPase的核苷酸交换,IC50为49.5 nmol · L-1。

图7 化合物34与GDP结合的人RalA蛋白的共晶结构(PDB ID: 6P0I)Figure 7 Co-crystal structure of compound 34 with GDP-bound human RalA (PDB ID: 6P0I)
6 结语与展望
共价抑制剂具有降低耐药性及靶向不可成药的靶点等优势,使其成为激酶抑制剂领域的焦点,并在疾病治疗中发挥着不可替代的作用,共价结合也早已应用到多个领域的研究当中[74-77]。但其同样具有潜在风险,如对合成速度缓慢且具有重要功能的靶蛋白的持续抑制可能会造成严重的副作用。为更好地发挥共价抑制剂优势,Bradshaw等[78]利用半胱氨酸反应性反式氰基丙烯酰胺亲电试剂鉴定了新的BTK、FGFR的共价抑制剂。开发具有低固有反应性弹头的共价抑制剂以降低脱靶可能性同样是共价抑制剂的发展方向之一,如McAulay等[79]于2020年发现了新的炔基苯并噁嗪和二氢喹唑啉亲电弹头,并开发出新型靶向半胱氨酸的JAK3共价抑制剂。除半胱氨酸外,研究人员也在努力开发共价靶向弱亲核氨基酸残基的共价抑制剂,如靶向谷氨酸和精氨酸[80-81]。共价抑制剂目前正受到广泛的关注,可以说这一浩瀚的宝库仍具有广泛的开发空间。相信随着众多开发与评价共价抑制剂的策略的提出与改进,共价抑制剂必将迎来蓬勃发展并为人类疾病的治疗带来新的希望。
猜你喜欢
中老年保健(2022年1期)2022-08-17
广西医科大学学报(2022年6期)2022-07-13
保健医苑(2022年5期)2022-06-10
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
医学概论(2022年4期)2022-04-24
中国典型病例大全(2022年7期)2022-04-22
中国药房(2022年7期)2022-04-14
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
