
基于微量合成的药物发现研究进展
2022-03-06 01:42陶昱岑徐淑静张续杰刘新泳展鹏
药学进展 2022年1期
陶昱岑,徐淑静,张续杰,刘新泳,展鹏
(山东大学药学院药物化学研究所 化学生物学教育部重点实验室,山东 济南 250012)
传统的药物发现来源于多轮的化合物设计、合成与生物活性评价,需要从超过1060种可能的化合物结构中去发掘潜在具有生物活性的小分子,该过程需要耗费巨大的人力、物力与时间,已经无法满足当前对新药快速发现的迫切需求,因此,新方法、新理念亟待发现[1]。
进入21世纪后,在高通量合成技术与生物活性筛选技术快速发展的基础上,药物发现的速度得到了极大的提升。其中,微量合成技术与生物活性筛选技术的有机结合极大地增加了先导化合物发现的概率与速率。微量合成相比于传统实验室合成具有极其显著的优点: 1)原料使用量大大降低,节约了实验成本的同时也降低了化学废料的排放量,更符合绿色化学的理念; 2)可以在短时间内平行合成大量的化合物; 3)微量合成辅以智能反应检测系统与合理的活性筛选方法,可以提高化合物从合成到活性筛选的自动化程度,极大地节省了人力物力。因此,微量合成技术在新药研发中的应用越来越广泛。微量合成技术的应用主要包括在以下几方面: 1)基于96或384孔微孔板的微量合成; 2)基于小分子或液滴微阵列的微量合成; 3)基于微流控芯片的微量合成; 4)靶标模板诱导的微量合成。本文精选近几年的研究实例,根据微量合成的不同类别,从药物化学的角度总结了基于微量合成的药物研究进展。
1 基于96或384微孔板的微量合成药物
通过在微孔板上快速构建具有结构多样性的化合物库,并辅以原位筛选技术可以极大地提高先导化合物的发现效率。选择一种合适的反应来生成具有结构多样性的化合物库是该策略的核心步骤,其基本特点为:目标化合物产率高、反应条件温和、反应易于操作。目前,该方法中常用的反应有点击反应[2]、酰胺缩合反应[3]、多组分反应[4]等。除此之外,也有文献报道了利用氨基与醛基生成亚胺的反应来快速构建聚焦型化合物库的方法等[5]。
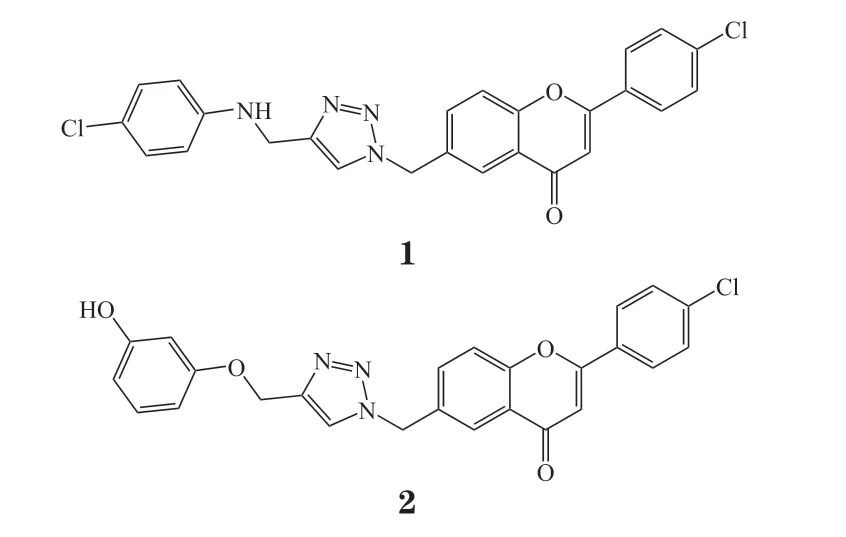
单胺氧化酶(monoamine oxidase,MAO)是一种具有多个结合位点的酶,主要分布于肝、肾、胰、心等器官。正常情况下,该酶可氧化胺类底物脱氨并产生过氧化氢。因此,MAO的过表达会使得机体内过氧化氢、乙醛等过量蓄积,从而导致神经元异常和情绪异常[6-8]。2016年,Jia等[9]利用点击化学微量合成辅以原位筛选技术,快速合成并筛选了40个黄酮衍生物。该化合物库设计思路为通过点击化学在黄酮C6位引入不同片段,伸入毗邻位点,从而实现潜在的双位点抑制作用,其中化合物1的活性最好(对MAO-A的IC50为 1.6 μmol · L-1;对MAO-B的IC50为2.1 μmol · L-1);化合物2的选择性最高,对MAO-A无活性,对MAO-B的IC50为(71.3±7.6)μmol · L-1,其选择性指数(SI)大于14。
溶酶体贮积症(lysosomal storage diseases,LSD)是一组遗传性代谢疾病,是由于基因突变致溶酶体中有关酸性水解酶缺陷,导致机体中相应的生物大分子不能正常降解而在溶酶体中贮积,引起细胞组织器官功能障碍[10]。由LSD引发的戈谢病在世界各地均有报道,然而现在临床对其缺乏有效的治疗手段。2017年,Martínez-Bailén等[11]在吡咯亚氨基糖骨架的基础上,通过点击化学在96孔板上快速构建了2类聚焦型化合物库,并通过后续生物活性筛选发现了选择性β-葡糖脑苷脂酶抑制剂3(IC50= 11 μmol · L-1)和4(IC50= 19 μmol · L-1)。2018年,Carmona等[12]通过点击反应原位筛选技术发现了α-岩藻糖苷酶抑制剂二聚体化合物5,β-半乳糖苷酶抑制剂二聚体化合物6,其中化合物5的抑制常数Ki为0.15 nmol · L-1;化合物6的Ki为5.8 μmol · L-1。
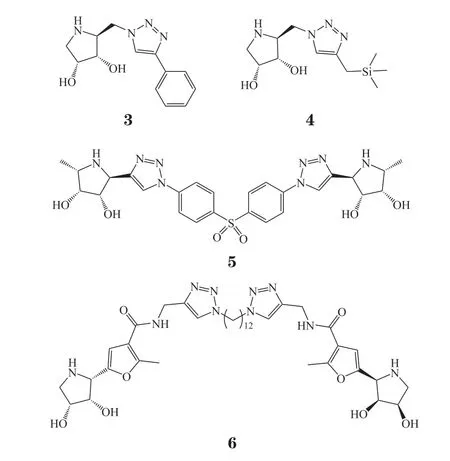
蓝氏贾第鞭毛虫是一种常见的能引起患者腹泻的寄生虫[13-15]。目前治疗贾第虫病的主要药物是硝基杂环类药物,包括咪唑衍生物、甲硝唑、替硝唑、噻唑、硝唑苯胺衍生物等。但这些药物的治疗失败率仍高达20%,并且有报道其体内外的耐药性[16]。Kim等[17]通过经典点击化学——铜(I)催化的炔烃-叠氮化物环加成反应[The copper(I)-catalyzed alkyneazide cycloaddition,CuAAC]快速微量合成了442个含有不同侧链取代基的氮杂环衍生物。对该化合物库直接进行活性筛选,发现化合物7对代表性的蓝氏贾第鞭毛虫突变株BRIS/83/HEPU/106具有良好的活性(EC50= 0.07 μmol · L-1)。
耐药性的产生是癌症治疗失败的主要原因之一。多药耐药相关蛋白1(multidrug resistanceassociated protein 1,MDR1)是一种腺嘌呤核苷三磷酸(adenosine-triphosphate,ATP)结合蛋白,其可以将药物泵出胞外,从而减少药物在肿瘤细胞中的蓄积,降低药物对肿瘤细胞的杀伤作用[18-19],抑制MDR1活性可以减少药物外排量,从而达到药物原有的治疗效果。据报道,天然产物中的黄酮类化合物是该靶标良好的小分子抑制剂,Wong等[20]选取黄酮结构为基本骨架,通过点击化学微量合成的方法在微孔板上高效地构建了含有300个二聚体黄酮衍生物的化合物库,原位筛选技术发现35个化合物对加有阿霉素(doxorubicin,DOX)的2008/MRP1细胞表现出较高的抑制率,后续活性复筛发现化合物8活性较好,EC50为(41±3) nmol · L-1,其对加有DOX的2008/MRP1细胞表现出了较好的活性,相比阳性对照MK571(9,EC50= 19 μmol · L-1)活性提升了400多倍。后续机制研究表明,化合物8在细胞内可以有效抑制MRP1细胞活性,从而逆转肿瘤细胞对DOX的外排作用,增强了DOX对耐药肿瘤细胞株的杀伤力。
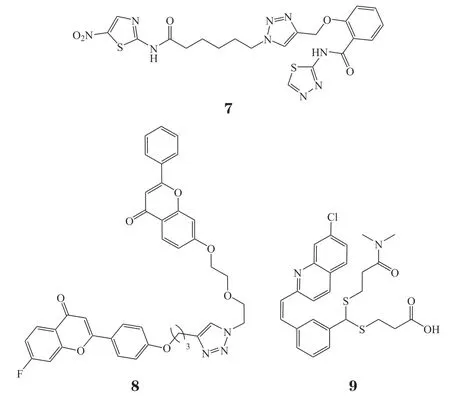
谷氨酰胺酶(glutaminase,GLS1)是一种氨基水解酶,是肿瘤细胞生长与增殖的重要调控蛋白,通过抑制GLS1活性可以有效抑制肿瘤细胞的增殖扩散[21]。因此,基于GLS1的小分子抑制剂研究非常重要。Xu等[22]以CB839(10)和BPTES(11)为先导化合物,通过生物电子等排策略,分别将CB839和BPTES右翼的苯环替换为三氮唑环。随后通过点击化学微量合成的方法将该片段与不同的小分子片段连接,在微孔板上高效地合成了200个目标化合物。后续活性筛选发现化合物12(IC50= 0.041 μmol · L-1)的活性超过阳性对照CB839(IC50= 0.11 μmol · L-1)。
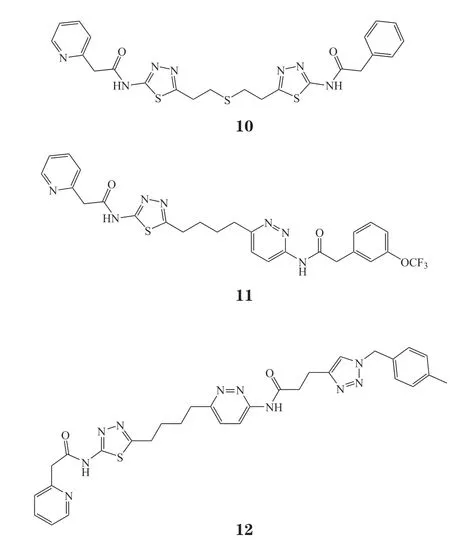
2018年,He等[23]选取O-苯并三氮唑-四甲基脲六氟磷酸盐(O-benzotriazole-N,N,N',N'-tetramethyl-uronium-hexafluorophosphate,HBTU)催化的酰胺缩合反应,在室温下仅用12 h便在微孔板中平行合成了4个化合物库(共768个化合物)。对库中化合物快速筛选发现化合物13 ~ 15活性较好,抑酶活性复筛发现化合物13 ~ 15对蛋白酪氨酸磷酸酶2(src homology 2 containing protein tyrosine phosphatase 2,SHP-2)具有良好的抑制活性,其中,化合物13的IC50为(1.5±0.1) μmol · L-1;化合物14的IC50为(1.4±0.04)μmol · L-1;化合物15的IC50为(2.3±0.04)μmol · L-1,化合物13的活性是阳性对照药物头孢磺啶[IC50=(16.8±2.0)μmol · L-1]的10多倍。药理机制实验表明,化合物13在细胞层面可以通过抑制SHP-2介导的信号通路来抑制肿瘤细胞的增殖。
近期,笔者所在课题组以细胞分裂周期因子25蛋 白(cell division cycle 25 phosphatase,Cdc25 phosphatase)磷酸酶的代表性抑制剂NSC663284(16)为先导化合物,选取优势萘醌片段,在其右翼通过点击化学微量合成的方式引入不同的取代基,发现了高活性、高选择性的Cdc25C磷酸酶抑制剂17,其 对Cdc25A的IC50为(0.53±0.03)μmol · L-1;对Cdc25B的IC50为(1.39±0.95)μmol · L-1;对Cdc25C的IC50为(0.09±0.01)μmol · L-1,其对Cdc25C活性相对于先导化合物NSC663284(IC50= 0.76 μmol · L-1)提高了近10倍[24]。此外,笔者所在课题组以二芳基嘧啶(diarylprimidines,DAPY)类化合物为先导,运用基于药效团模型与靶标结构的药物设计对DAPY类非核苷类逆转录酶抑制剂(non-nucleoside reverse transcriptase inhibitor,NNRTI)进行结构优化[25]。随后通过点击化学微量合成的方法快速合成了234个DAPY衍生物,辅以原位筛选技术与活性复筛,短时间内发现了人类免疫缺陷病毒1型(humam immunodeficiency virus-1,HIV-1)抑制剂化合物18,其EC50为3.28 nmol · L-1。同时,化合物18对HIV双突变株RES056也具有抑制活性,其EC50为481 nmol · L-1。
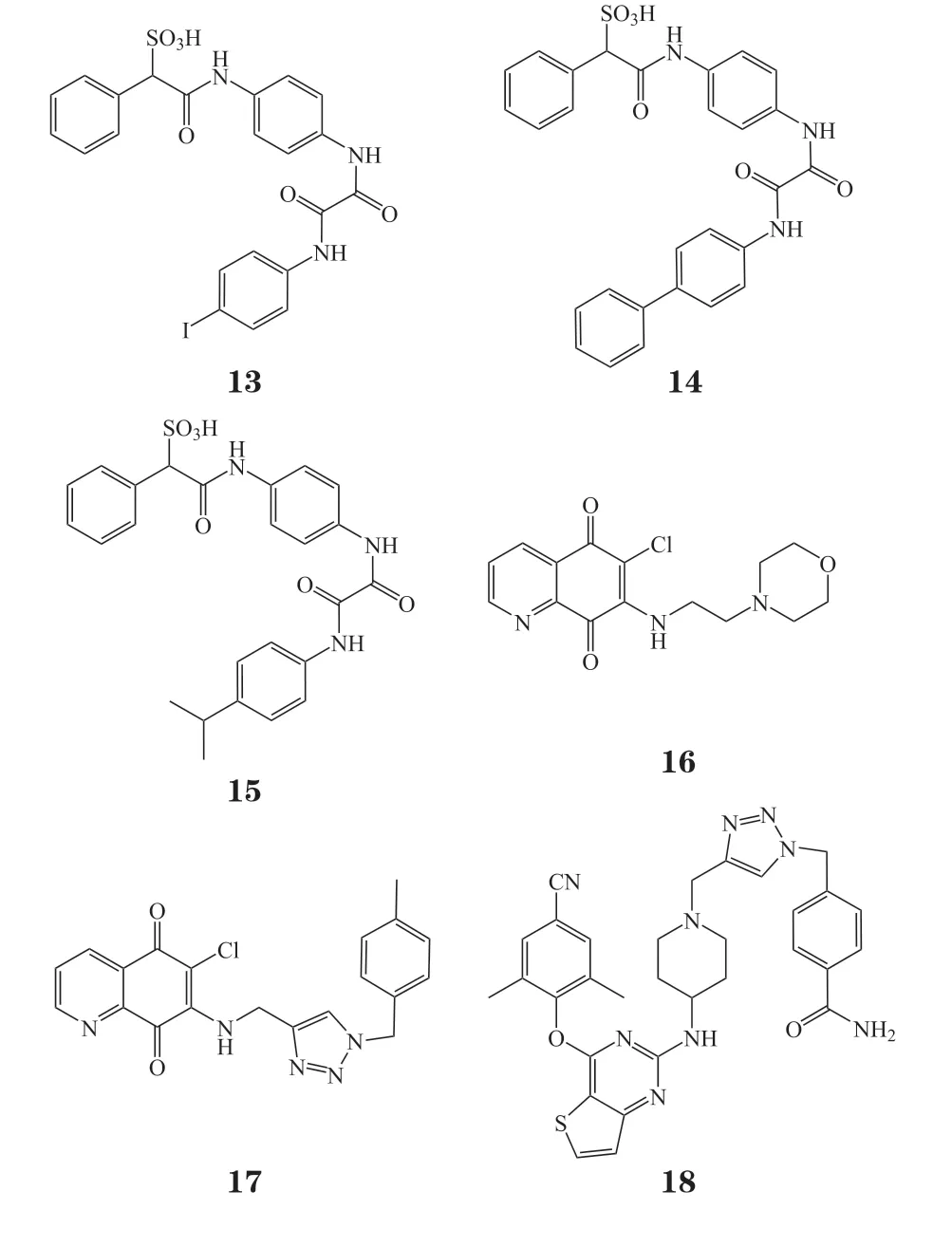
2021年,Sangouard等[26]将声学液滴喷射(acoustic droplet ejection,ADE)技术与基于微孔板的微量合成相结合,短时间内构建了大型组合大环化合物文库。随后通过直接筛选快速发现了鼠双微粒体基因-2蛋白和p53蛋白(murine double minute 2-p53,MDM2-p53)蛋白-蛋白相互作用的抑制剂19,其展现出了良好的抑酶活性,IC50为(0.6 ±0.2) μmol · L-1。传统的基于微孔板的高通量合成受到高成本移液头和合成所需的毫克级前体化合物等因素的限制。为克服上述缺点,Sangouard等[26]对建库方法进行了改进,并首次报道了应用ADE技术,将砌块库逐步转移到384孔微量滴定板中来合成大型组合大环化合物文库的方法。该方法与传统微孔板合成(反应液体积为微升)相比,仅需要纳升体积的反应液,为基于微孔板的微量合成提供了全新的思路,避免了前体物料的过度消耗,进一步实现了绿色化学的理念。
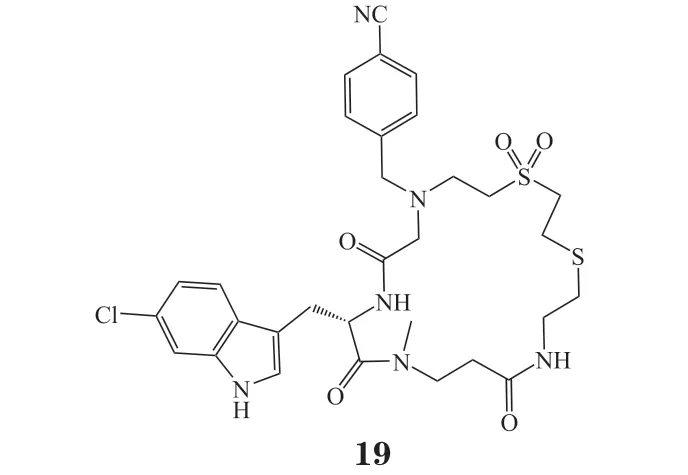
需要指出的是,CuAAC是经典的点击化学反应之一,但由于该反应中催化所必需的铜离子对生物体系中的酶、细胞等具有毒性,因此易造成假阳性或假阴性结果。2004年,Agard等[27]提出了无铜催化的炔烃-叠氮环加成反应(strain-promoted azidealkyne cycloaddtion,SPAAC)。通过环的构象限制,可以提高炔烃的反应活性,从而使得SPAAC可以在温和的无铜催化下进行。半胱氨酸蛋白酶(Caspase)是一类非常保守的蛋白酶,其可以调控细胞的凋亡和促炎细胞因子的成熟,Qian等[28]选择Vertex公司研发的口服Caspase-1抑制剂pralnacasan(20)为先导化合物,通过骨架跃迁策略,将pralnacasan分子中的六元和七元并环结构替换为三氮唑并八元环,而后通过SPAAC反应,在微孔板中快速合成了52个化合物。由于该反应液中不含铜离子,因此可以直接用于细胞活性的初步筛选,接着选取抑制率最高的5个化合物对其进行了后续的活性复筛。生物实验结果表明化合物21活性较好,其对Caspase-1的IC50为(0.899±0.001) μmol · L-1、对Caspase-3的IC50大 于 100 μmol · L-1、对Caspase-7的IC50大于140 μmol · L-1。
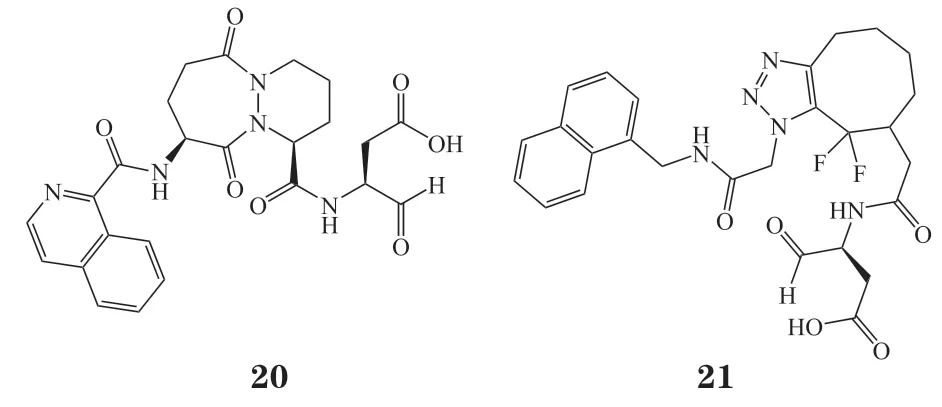
在96或384微孔板上进行的微量合成除了上述的点击反应外,还包括多组分反应。2020年,Osipyan等[29]报道了利用微量多组分反应制备亚胺基吡咯烷聚焦库的方法。该方法核心是利用即时喷点移液(immediate drop on demand technology,I-DOT)技术将载有原料的溶液准确加入到384孔板中,并控制每孔所含反应液为660 nL。反应16 h后,将反应液注入质谱(mass spectra,MS)中进行检测,结果显示大部分微孔(66%)中目标产物的产率都在中等及偏上水平(≥ 50%)。接着,Osipyan等[29]又随机选择了12个微孔中的反应进行毫摩尔级的扩大量合成以验证其产率。结果显示,反应底物有环酮时,反应具有不错的产率(82% ~ 83%),然而当酮类底物为环丁酮时,反应产率仅有34%。醛基底物中,脂肪醛相比于芳香醛,更有利于目标化合物的生成。该方法的报道丰富了微孔板合成技术的反应类型,为后续基于微孔板的微量合成提供了新的反应类型。同时,I-DOT技术的出现使得微量合成反应中加料量的精确度进一步提升,更加有利于微孔板中反应的进行。
2021年,Gao等[30]通过声学传感移液技术将3组分反应液快速固定在微孔板中,短时间内合成了1 536个化合物,后续通过原位筛选和抑酶活性复筛实验发现化合物22,其对多发性内分泌腺瘤1型基因编码蛋白(Menin)表现出了微摩尔级的活性,其IC50为2.8 μmol · L-1。除此之外,Gao等[30]通过共晶进一步探讨了化合物22与Menin蛋白的结合模式(见图1A)。2021年,Sutanto等[31]通过Ugi多组分反应和Passerini多组分反应分别在96和384微孔板上快速构建了具有结构多样性的化合物库,由于终产物水溶性较差,当反应完成时,大部分终产物在微孔中直接析出,核磁氢谱显示,该类析出化合物纯度较高(纯度大于 80%),而未析出的化合物可以通过快速柱层析迅速纯化。随后的高通量蛋白-小分子共晶实验发现,4个苗头化合物23 ~ 26与活性位点的氨基酸残基Cys145形成共价结合,通过荧光共振能量转移(fluorescence resonance energy transfer,FRET)实验测定以上4个苗头化合物对2019新型冠状病毒(SARS-CoV-2)的主蛋白酶(3CL-Pro)的活性,其IC50分别为3.96、139.1、9.39及2.36 μmol · L-1,其结合模式如图1B所示。
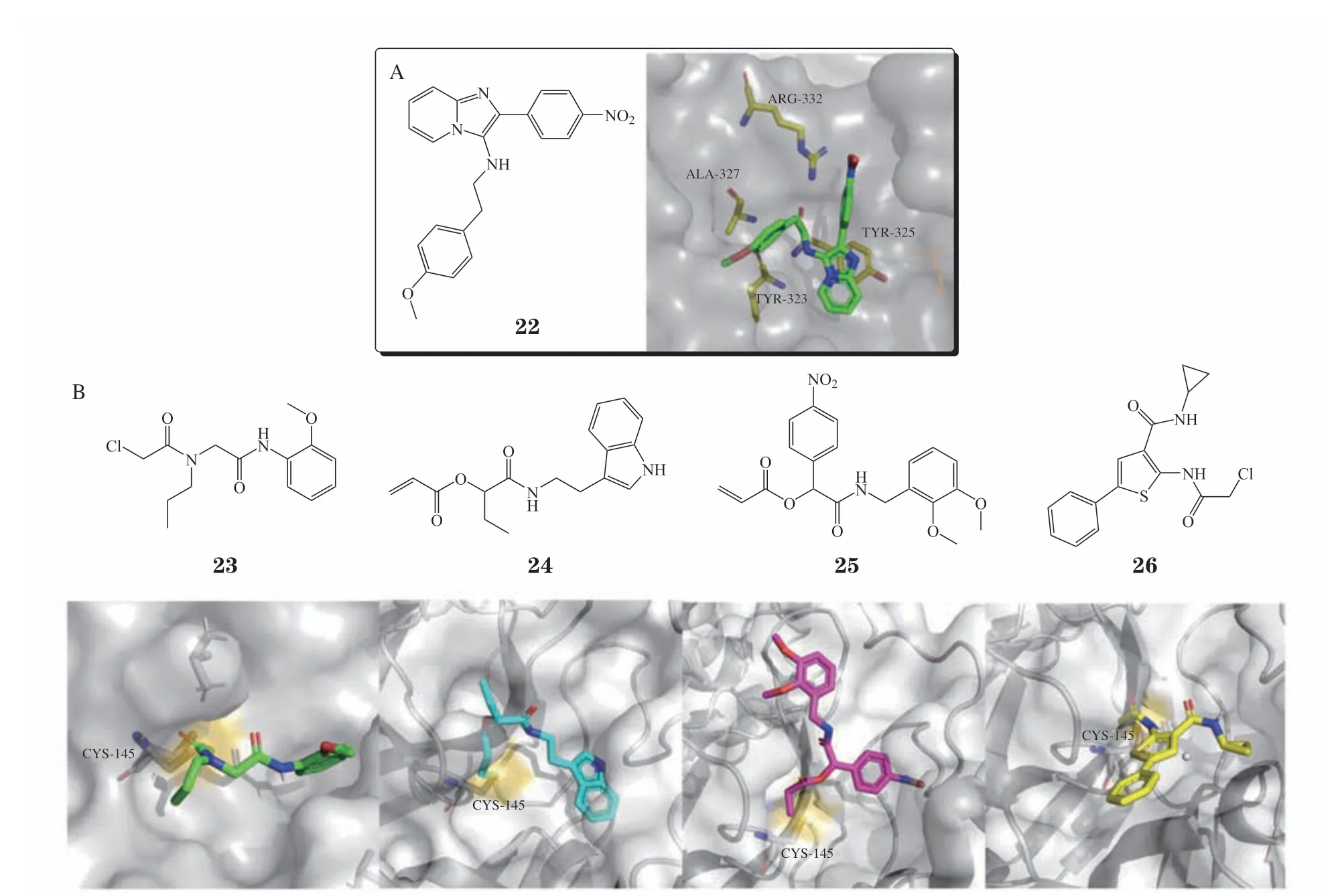
图1 化合物22 ~ 26的结构式及其与靶蛋白的结合模式Figure 1 Structures and binding modes of compounds 22 ~ 26
综上,基于微孔板的高通量合成技术可以在短时间内平行合成大量化合物,极大地提高了苗头化合物/先导化合物发现的概率。同时,微量的条件下,反应溶剂及试剂的消耗量极小,节约原料的同时,也减少了有毒废弃物的排放,更符合绿色化学理念。除此之外,基于微孔板的微量合成技术由于其操作的简便性、方法的实用性等优点,越来越多的实验室将该方法运用到了新药研发中[32-33]。
2 基于小分子或液滴微阵列的药物
1995年,Schena 等[34]发明DNA微阵列技术,将已知序列的寡核苷酸固定在固体表面上,同时将待检测的探针核酸序列与上述已知的核苷酸序列进行杂交,从而实现基因信息的快速检测。随着化学的发展,微阵列技术不再局限于DNA分子,蛋白质分子、化学合成小分子等也相继被固定到固相表面用于微阵列分析。小分子微阵列技术(small molecule microarray,SMM)是指将目标化合物有序地固定到固体表面,从而实现快速、平行的活性筛选的技术[35]。SMM技术包含:化合物库的合成、化合物的固定等,与传统有机溶剂中的合成相比,SMM技术中的固相合成技术具有以下显著优点:自动化程度高、反应产率高,因此,其应用越来越广泛[36-38]。
乳腺癌相关基因1编辑蛋白(breast cancer 1 protein,BRCA1)是一种包含有磷酸化丝氨酸结合域的细胞调控蛋白,其可以抑制细胞的癌变,在DNA损伤的修复中发挥重要作用。在多种肿瘤细胞中均发现了突变型的BRCA1,因此针对BRCA1突变型的小分子抑制剂研发非常重要[39]。Na等[40]通过SMM技术,将优势氨基酸片段固定于活化玻璃板上,通过微量的酰胺缩合反应,快速合成了101个拟肽类化合物。接着Takaoka等[39]使用N,N-二甲基甲酰胺和二氯甲烷对玻璃板进行多轮的清洗,快速除掉残留的反应液和催化剂,从而对玻璃板上的化合物进行准确的活性筛选。该方法极大地缩短了化合物合成到先导化合物发现的时间,Takaoka等[39]利用该方法成功地发现BRCA1抑制剂27(IC50= 0.31 μmol · L-1)(见图2)。
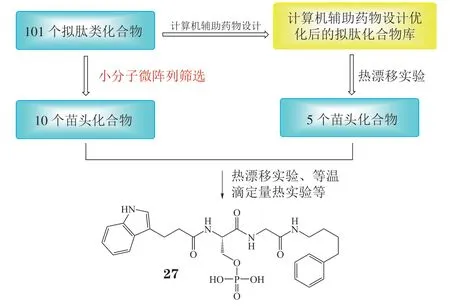
图2 先导化合物27的发现过程及结构式Figure 2 Discovery and structure of compound 27
2017年,Peng等[41]针对多聚ADP-核糖聚合酶(poly ADP-ribose polymerase,PARP)进行合理药物设计,利用SMM技术,通过CuAAC点击反应在树脂板上微量合成了1 120个化合物,后续的活性筛选发现化合物28是PARP14双位点抑制剂(IC50= 0.49 μmol · L-1)。为了探讨化合物28与PARP14的具体结合模式,Peng等[41]进行了共晶的培养,结果显示化合物28同时结合于2个空腔中,并与Ser1700、Ser1688、Thr1684、Ser1722等多个氨基酸残基形成了氢键作用(见图3)。
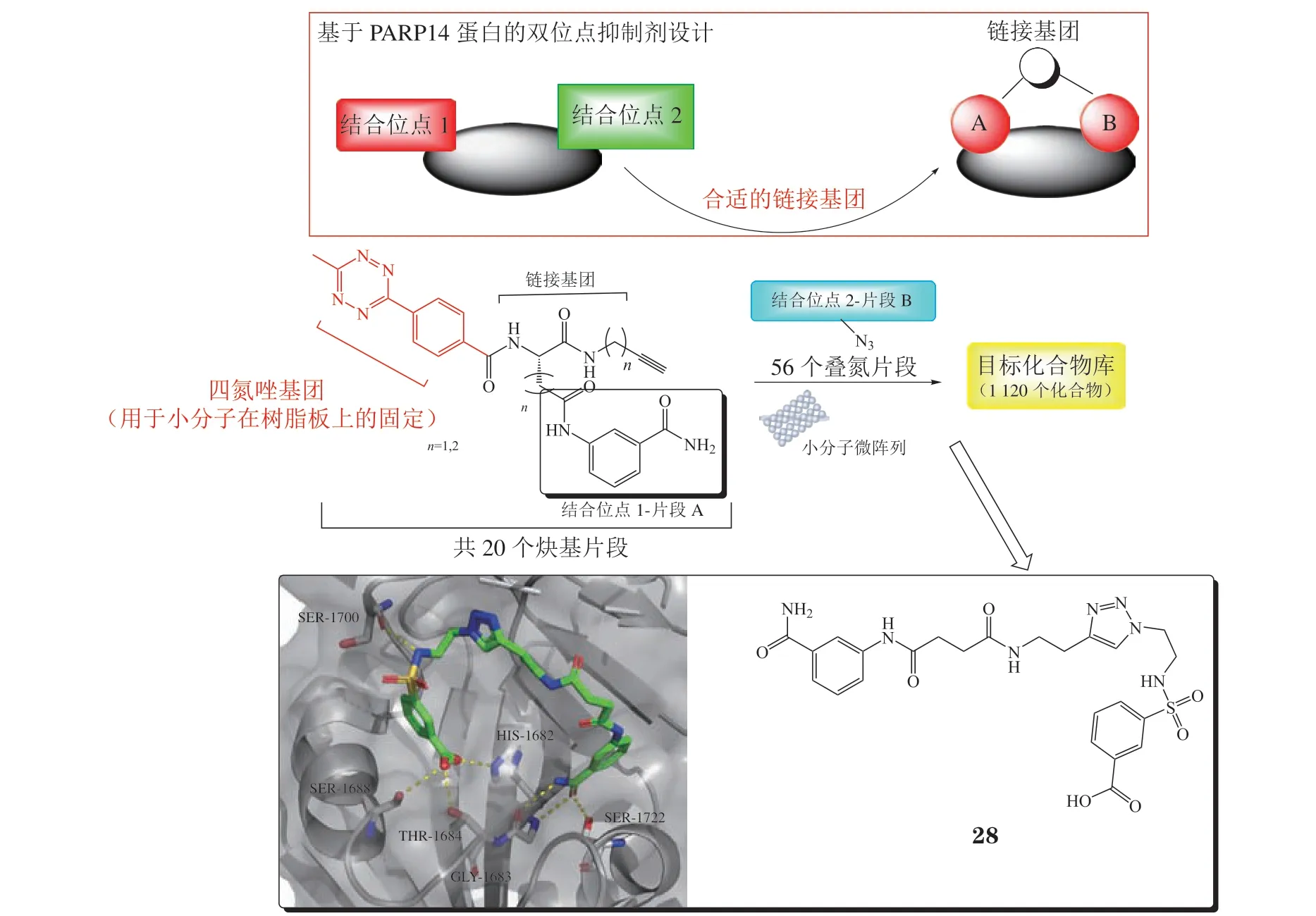
图3 化合物28的设计、合成过程及其与PARP14蛋白的结合模式图(PDB ID:5LYH)Figure 3 The design and synthesis of compound 28 and its mode of binding with PARP14(PDB ID:5LYH)
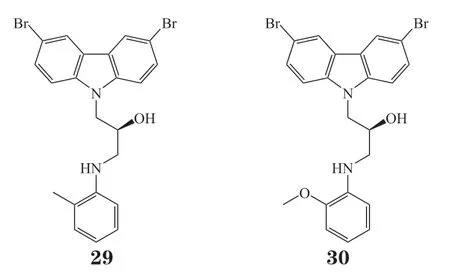
RNA是体内重要的遗传信息载体,其中微小核糖核酸21型(microRNA-21,miRNA-21)在多种肿瘤中发现了过表达的现象。敲除肿瘤细胞中的miRNA-21后可以诱导肿瘤细胞的凋亡。2010年,Medina等[42]通过小鼠模型确证了miRNA-21的过表达与前体B细胞淋巴瘤的发生直接相关。然而,针对RNA的高亲和力小分子抑制剂鲜有报道。2017年,Connelly等[43]通过微量合成将20 000个小分子固定于γ-氨基丙基硅烷 (γ--aminopropyl silane,GAPS)玻璃板上,接着将其与荧光标记的premiRNA-21共孵育1 h后,通过荧光成像的不同判断小分子与miRNA-21的结合力强弱(评分Z-score>3即被认为是苗头化合物)。其中,19个化合物(Z-score>3)表现出了对miRNA-21较好的亲和力。后续活性复筛发现化合物29和30活性最好,其中,化合物29的解离常数(Kd)为(2.3±0.5) μmol · L-1;化合物30的Kd为(0.8±0.2)μmol · L-1。
2019年,Rosenfeld等[44]报道了在纳米聚合材料上微量合成多肽的方法,其开发的纳米聚合材料上有序地分布着亲水性微孔,孔内含有活化的光敏性链接基团,从而有效地支持孔内肽段的固相合成。Rosenfeld等[44]还证实该方法除了支持肽段合成以外,还可以在微孔中加入反应液进行微量的小分子化合物合成。后续通过特定波长的紫外光照射光敏性链接基团,使其断裂,从而释放出目标产物。通过改变光线照射时长与光线强度等因素,可以有效控制目标化合物释放的数量和时间等。该技术实现了目标化合物的微量合成与光诱导的控制释放,辅以后续的原位筛选技术可以极大地提高化学合成到活性筛选的效率,增加苗头化合物的发现机率。
除了上述小分子微阵列技术以外,液滴微阵列也同样是一种常见的微阵列技术。液滴微阵列技术是指将含有目标化合物的甘油溶液有序滴加在固相表面,从而直接进行活性筛选的技术。该技术有着诸多优势: 1)无需将溶液蒸干,液滴即为合成反应的微环境; 2)与小分子微阵列相比,液滴微阵列技术无需通过化学反应将化合物共价结合于平板表面,甘油自带的表面张力与附着力保证了液滴有序地排列在固相表面; 3)甘油的微环境可以使得多种酶在体外环境中变得稳定,同时,甘油也是一种极佳的液体介质,其与二甲亚砜、水等常见溶剂完全互溶[45-47]。
由于干细胞的自我更新能力和高度分化能力[48-49],使其在过去的几十年中引起了医学生物学家们越来越广泛地关注。通过基于干细胞的高通量筛选发现对自身组织具有促进更新与修复能力的小分子化合物是一项极有意义的研究[50-52]。但该研究的实施面临着诸多问题,其中最主要的问题是干细胞在培养时会自发地细胞分化,从而使得实验失败。2017年,Tronser等[53]证明了液滴微阵列技术可以成功阻止干细胞的自发性细胞分化。实验中,Tronser等[53]发现液滴中的鼠源干细胞状态趋于稳定,自发的细胞分化作用停止了72 h。后续机制实验表明,该现象可能归因于固相载体表面的孔隙率和粗糙程度。虽然该实验仅短时间内抑制了干细胞的自发分化作用,但为后续干细胞药物研发提供了全新的思路与方法。
2020年,Brehm等[54]在之前研究基础之上报道了光控释放目标产物的液滴微阵列技术。他们在玻璃薄片上覆盖一层多孔的纳米聚合物材料,该材料每个微孔中都含有1个光敏连接链用以诱导微孔中的反应进行。当反应完成时,通过多次洗涤该玻璃板,将残留的反应液清除干净,之后在纳米微孔中滴加溶剂,并同时使用365 nm的紫外光照射来控制产物按需释放入微孔液滴中。后续通过在液滴中直接加入细胞,从而进行快速的活性筛选。Brehm等[54]在该技术基础之上,使用Ugi四组分反应,在微滴中快速合成了588个化合物。
3 基于微流控芯片的药物
从中世纪的炼金术开始,人们习惯于在玻璃器皿(烧瓶、烧杯)中操纵反应原料,获得产物。然而,面对人类对新物质、新规律的不断需求,传统玻璃器皿的劣势日渐凸显:集成化、自动化程度低,资源浪费,选择性、重现性差,以及安全隐患等。在此背景下,微流控芯片反应器应运而生,并以其独特优势,在合成方法学、药物筛选以及大规模应用等诸多领域扮演着重要角色。
微流控芯片反应器一般是指微加工和精密加工技术制造的小型反应系统,内部微通道尺寸在亚微米到亚毫米数量级。常见的微流控芯片反应器从材质上分,有玻璃/石英/硅芯片、聚合物芯片和金属芯片3类[55-57]。
玻璃和石英芯片因具有优异的电渗、光学和表面性质,其刻蚀加工方法和表面改性的化学方法比较成熟[33,58]。但由于玻璃芯片不耐强酸,因此其应用也具有一定局限性。
有机聚合物类芯片由于其种类较多、加工方便,是玻璃和石英芯片不错的替代品。聚二甲基硅氧烷(polydimethylsiloxane,PDMS)芯片具有良好的光学性能,价格便宜易加工,应用比较广泛。PDMS的化学性质大部分情况趋于稳定,但遇到丙酮等会发生形变,并且这类芯片散热不及玻璃和石英[59]。
不锈钢芯片相比于以上3种芯片的稳定性较高。这类芯片一般为多模块组装而成,易拆卸可清洗,避免了微通道发生堵塞而使整个反应器报废。但由于该类芯片的特殊材质和各模块的精密加工,不锈钢芯片成本非常高,对微加工技术的要求也很高[60-61]。
不同材质的微流控反应器都具有扩散距离短、传质更快的优点。化学反应的实质在于分子的碰撞从而反应生成新的化学键,因此合成中反应物的混合情况是提高反应效率的关键之一。其次,微流控芯片反应器相比传统圆底烧瓶等反应容器在比表面积上有了飞跃。微流控芯片反应器比表面积通常可以达到5 000~50 000 m2· m-3,超大的换热面积使微反应器的热传导系数与常规反应器相比高出10倍,无论是对反应的加热还是反应本身释放的热量都可以快速传递,从而使反应温度更稳定,反应过程的控温更容易[62]。
以上几大优势使得微反应器在有机合成中的应用越来越广泛,这方面的研究也在不断深入。
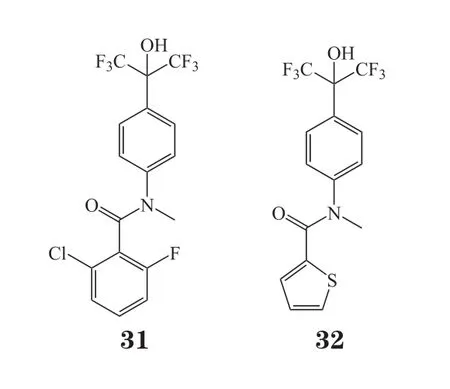
Grisoni等[63]将人工智能辅助药物设计与微流控反应器有机结合,极大地提高了苗头化合物发现的效率。他们首先针对肝X受体(liver X receptor,LXR)利用人工智能深度学习的方法对所获得化合物库进行虚拟筛选,同时对筛选所得分子进行合成可行性分析,最后得到了44个具有全新结构的可合成化合物。其中,41个化合物通过自动化的微流控反应仪合成,3个化合物从试剂公司购买所得。接着Grisoni等[63]对其进行了活性测试,发现化合物31和32分别是LXRα和LXRβ型的激动剂,其EC50分别为 (0.183±0.006)和(0.34±0.02) μmol · L-1。该结果进一步证明了微流控合成在药物合成领域的可行性。同时,将微流控反应仪与人工智能深度学习、液相色谱-质谱联用技术(high performance liquid chromatography-mass spectrometry,HPLC-MS)等分子设计与反应实时监测的方法整合,加快了苗头化合物发现速率。
4 基于靶标模板诱导的合成策略
靶标模板诱导的合成(target-guided synthesis,TGS)是药物发现中一种极其重要的策略,该策略以蛋白质靶标为模板诱导与其结合的小分子片段相互连接。该策略主要分为2类: 1)动态组合化学(dynamic combinatorial chemistry,DCC),其目的在于建立可变通的适合新药开发的组合库,其途径类似于分子水平上的达尔文进化论。通过“适者生存”的方法,在组合库重组的过程中,与模板无关的化合物或结合比较弱的配体逐渐“死亡”减少,解离成原来的“分子单元”,而这些分子单元在模板的引导下重新组合、反应,形成新的活性配体,它们以此方式“繁殖”增加。组合库的分子单元作为“生存”的竞争者,依赖于单元之间的可逆反应,与模板牢固结合的分子将产生新分子,然而,与模板结合较弱的分子将作为原料留在母液中。2)不可逆的蛋白模板诱导合成(kinetic target-guided synthesis,KTGS),该方法与DCC唯一不同的是,KTGS中的片段链接是不可逆的[64-66]。目前,2种方法均已被成功运用在多种靶标的苗头化合物的发现中[67]。
2017年,Wang等[68]在DCC策略的指导下,通过点击化学原位合成技术将低活性(IC50>1 mmol · L-1)的不同小分子片段拼接,发现了2个具有透膜性的化合物33和34(见图4A),其IC50分别为139和66.7 μmol · L-1。同年,Bhardwaj等[69]以2型环氧化酶(cyclooxygenase-2,COX-2)蛋白为模板,通过原位点击反应发现化合物35和36(见图4B)(化合物35:IC50= 0.09 μmol · L-1;化合物36:IC50= 0.05 μmol · L-1)展现出了与阳性药物塞来昔布(IC50= 0.07 μmol · L-1)相当的抑酶活性。后续动物模型实验发现化合物35和36在小鼠体内活性明显优于阳性药物塞来昔布,其半数有效量(median effective dose,ED50)分别为0.44 和0.12 mg · kg-1,而塞来昔布ED50为10.8 mg · kg-1。构效关系分析表明,在引入甲磺酰基药效团后,化合物的活性有所提高。
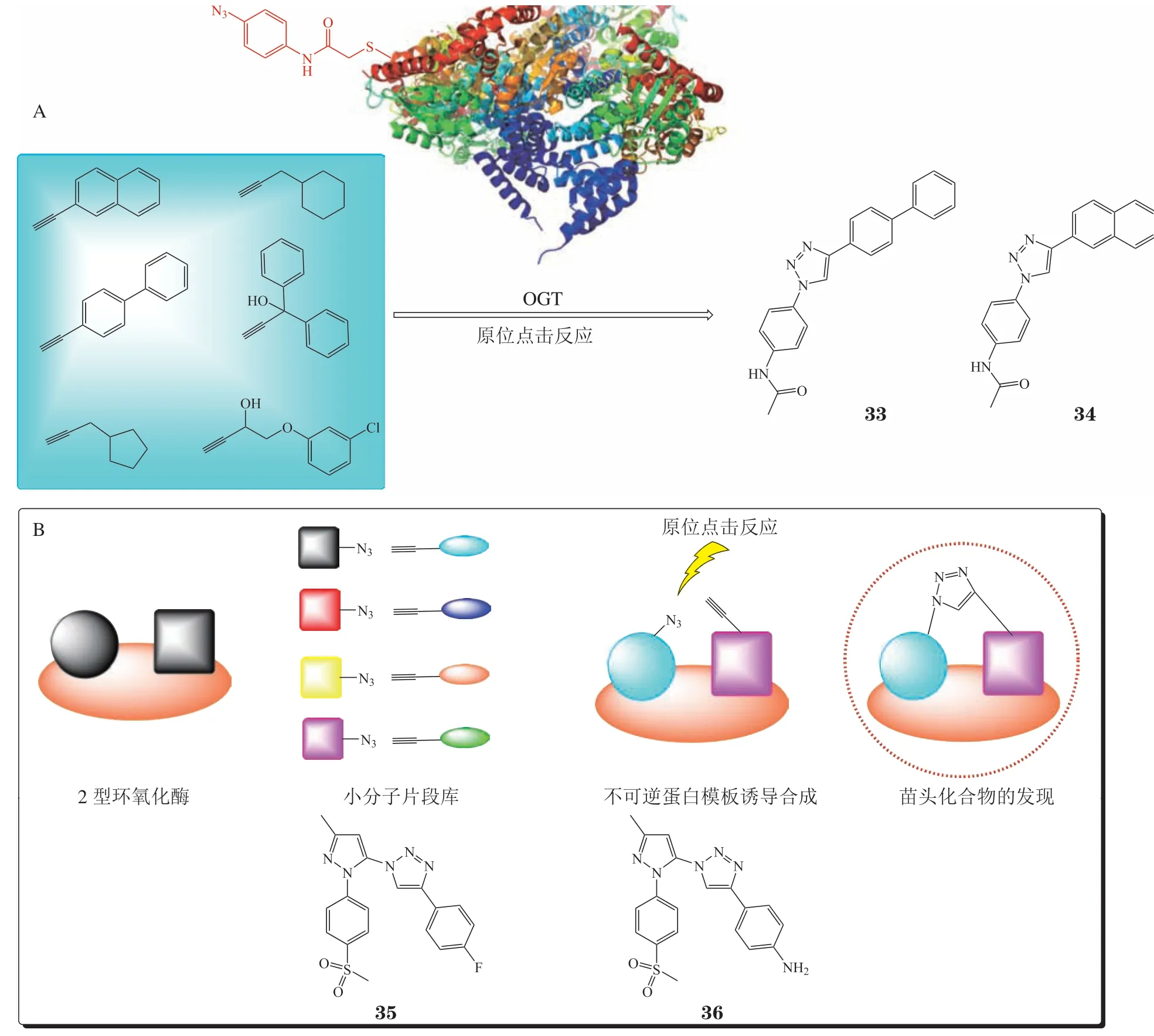
图4 化合物33 ~ 36的发现过程及其结构式Figure 4 The discovery and structures of compounds 33 ~ 36
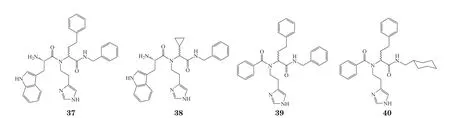
靶标模板诱导的合成中除了常见的点击反应以外,2020年,Mancini等[70]首次报道了蛋白模板诱导的原位四组分Ugi反应。Mancini等[70]利用endothiapepsin蛋白为模板,选取羧基砌块库、氨基砌块库、氰基砌块库和醛基砌块库合成了48个化合物,其中化合物37和38对endothiapepsin蛋白表现出了微摩尔级的活性,其IC50分别为(1.3±0.03)和(3.5±0.1) μmol · L-1。随后,Mancini等[70]又将原位四组分反应成功应用在了细菌β-滑动钳DnaN蛋白模板上,并发现了化合物39和40可以与靶标蛋白相结合,其Kd分别为650和510 μmol · L-1。原位四组分反应在2种蛋白模板上的成功丰富了TGS策略中的反应类型。除此之外,Mancini等[70]观察到Ugi反应中的副反应(Pictet-Spengler环合反应)也在原位合成实验中同时发生,因此,基于该类副反应的蛋白模板诱导合成或许又是一种全新的尝试。
值得注意的是,在大多数靶标模板诱导的小分子片段组装中,对酶的纯度要求较高,这也限制了该技术的广泛应用。2018年,Antti等[71]报道了在细胞内的蛋白模板诱导合成,为那些难以纯化的蛋白模板提供了新的思路。
综上所述,虽然TGS展现出了其在苗头化合物发现中的高效性,但该技术依旧存在诸多限制。比如对蛋白质与目标小分子复合物的分析技术还未完全成熟,虽然现阶段可通过共晶、核磁共振以及HPLC-MS等技术对蛋白质-小分子复合物进行分析,但这几类技术需要耗费大量的时间。除此之外,文献中报道的能用于TGS方法的反应比较匮乏,常见的反应有:点击反应、酰胺缩合反应、环氧乙烷的开环反应等。因此,新的反应类型和新型检测技术亟需被发现。
5 结语与展望
在新药研发中,药物化学家们通过微量合成将优势片段、优势结构进行重组装,在保证药物设计合理性与结构新颖性的同时,加速了化合物库的构建,解决了新药研发中“如何快速获得大量目标化合物”的难题。随着分析检测技术的发展,构建化合物库的同时也能对其进行结构鉴定,如微量合成与LC-MS技术的联用等。
同时,在多样性导向合成理念的指导下,通过微量合成将现有小分子砌块库排列组合,进一步丰富了化合物库的结构多样性,为药物化学家们阐明靶标与化合物的潜在构效关系提供了有力保障。除此之外,生物活性筛选技术的发展,使得对化合物库的活性筛选更加准确,假阳性/假阴性结果逐步减少,进一步促进了苗头化合物的发现。在苗头化合物的基础之上,微量合成与晚期多样性修饰等新理念的有机结合可以缩短从苗头到先导化合物所需的时间。
诚然,微量合成加速了苗头化合物发现的速率,但以下问题依旧值得注意: 1)片段的类药性; 2)小分子砌块库的多样性; 3)分子片段的手性异构以及其大小等,这些问题直接决定了微量合成所构建化合物库的质量,从而间接决定了最后苗头化合物发现的成功与否。此外,微量合成所使用的反应类型也是库的构建中一项决定性因素。因此,基于微量合成的药物发现需要药物化学家们综合考虑多种因素[72-73]。
综上所述,微量合成与新方法、新理念的创新性组合,帮助药物化学家们节约了大量的人力和物力,缩短了药物研发所需的时间,将“以小博大”体现得淋漓尽致。虽然现阶段微量合成在药物研发中依旧面临着部分不足之处,但随着各学科的发展,学科之间的交叉与融合越来越广泛、新方法与新理念的有机结合越来越普遍,微量合成在新方法、新理念的促进下变得越来越成熟,为应对未来公共健康危机的新药研发提供了全新的思路。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
中国动物保健(2022年2期)2022-05-05
现代计算机(2020年17期)2020-08-10
中国建筑金属结构(2019年4期)2019-05-15
农业与技术(2018年12期)2018-11-09
环球市场信息导报(2018年1期)2018-05-30
中国美容医学(2017年6期)2018-02-05
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
