
靶向蛋白非催化功能的药物开发新策略
2022-03-06 01:42杨欣语宋宜辉余斌
药学进展 2022年1期
杨欣语,宋宜辉,余斌
(郑州大学药学院,河南 郑州 450001)
蛋白质在与生理功能密切相关的细胞信号转导中发挥着重要作用。完整的结构域是蛋白质发挥正常生理功能的结构基础。一个经典的蛋白酶常由保守的催化结构域和非催化结构域组成。催化结构域与酶的催化活性直接相关,而非催化结构域则主要通过催化功能非依赖的方式如支架蛋白功能、变构调控和蛋白-蛋白相互作用等确定底物的特异性,或协调其与信号通路不同组分之间的相互作用,进而调控酶的活性[1-2]。鉴于蛋白酶催化功能的重要性,长期以来靶向蛋白酶保守的催化功能开发抑制剂一直是药物研发领域的热点。但这类抑制剂却面临着选择性不高、特异性不强和临床副作用多等挑战,而蛋白非催化功能的特异性为高选择性抑制剂的开发提供了研究方向,也日渐引起了大家的关注。基于蛋白质非催化功能的最新研究进展和抑制剂开发策略,本文通过系统总结靶向蛋白非催化功能开发抑制剂的新策略,即非催化结构域策略、变构调控策略和蛋白-蛋白相互作用策略,为靶向蛋白非催化功能的抑制剂开发提供研究思路。
1 蛋白非催化结构域策略
酶的非催化结构域可作为支架蛋白,通过与信号通路中的其他组分结合,在细胞信号传导中发挥着关键作用,如胺氧化酶蛋白中的赖氨酰氧化酶样蛋白-2(lysyl oxidase-like 2 protein,LOXL2)N末端特有的富含半胱氨酸的清道夫受体(scavenger receptor cysteine-rich,SRCR)结构域可通过非依赖催化作用的支架功能调节内皮细胞外基质和血管的生成[3]。研究表明,蛋白质非催化结构域参与调控RNA代谢,DNA修复,细胞分裂、分化和基因组稳定性等各种细胞过程,与诸多疾病如癌症和血栓的发生密切相关[4-7]。与传统的靶向蛋白保守催化功能的药物不同,针对蛋白特异性非催化结构域开发的小分子抑制剂可能具有更高的选择性,在疾病治疗领域具有较大的应用前景。目前已成功靶向了多个蛋白的非催化结构域,如赖氨酸特异性去甲基化酶4A(lysine-specific demethylase 4A,KDM4A)的TUDOR结构域和局部黏着斑激酶(focal adhesion kinase,FAK)的非催化黏着斑定位区(focal adhesion targeting,FAT)等[6,8],部分代表性的抑制剂如表1所示。

表1 蛋白非催化结构域策略相关靶点研究现状Table 1 Current status of research on targets with non-catalytic domain strategy
1.1 靶向赖氨酸特异性去甲基化酶4A的非催化结构域研究现状
在人类基因组中,含有Jumonji C结构域的组蛋白去甲基化酶(Jumonji C domain-containing histone demethylase,JHDM)家族成员组蛋白赖氨酸特异性去甲基化酶4(histone lysine-specific demethylase 4,KDM4)包 含A、B、C、D和E 5种不同的亚型,可以催化组蛋白H3第9位赖氨酸(histone H3 lysine 9,H3K9)和组蛋白H3第36位赖氨酸(histone H3 lysine 36,H3K36)的去甲基化[9-12]。5种亚型KDM4的N端均含由Jumonji N和Jumonji C组成的保守催化结构域,分别为JmjN和JmjC,但C末端差异较大,仅4A、4B和4C 3种亚型的C末端包含独特的串联植物同源结构域(plant homeodomain,PHD)和TUDOR结构域(PDB ID:5D6X)[6]。研究表明:KDM4A、4B和4C的过表达与各种癌症的发生发展密切相关,是抗癌药物开发的重要作用靶点[13-15]。目前报道的KDM4小分子抑制剂主要靶向其保守的催化结构域,根据其作用机制可分为3大类: 1)金属螯合抑制剂,如N-草酰甘氨酸(N-oxalylglycine,NOG)是一类α-酮戊二酸(α-ketoglutarate,α-KG)辅助因子的模拟物,通过与酶催化位点的Fe(Ⅱ)分子竞争性地结合而抑制KDM4蛋白的酶活性[16]。但这类化合物的亲水性结构往往导致其具有较差的细胞穿透能力,因而限制了其临床应用[17]。2)金属辅助因子干扰物,如双硫仑(disulfiram)和依布硒(ebselen),通过阻止辅助因子如铁和锌的结合而抑制KDM4的酶催化活性[13]。3)组蛋白底物的竞争性抑制剂,如MS275,其通过与底物组蛋白的竞争性结合而达到抑制效果[13]。但由于KDM4酶家族催化机制的保守性,这些抑制剂面临着选择性差的问题。研究发现,含有TUDOR结构域的蛋白质在调节细胞过程,如RNA 代谢、DNA损伤反应和染色质修饰中也发挥着至关重要的作用。因此,靶向KDM4中独特的非催化TUDOR结构域也成为开发高选择性抑制剂的潜在研究方向[4-5]。KDM4A也称为含Jumonji结构域蛋白2A(Jumonji domain containing 2A,JMJD2A),在调节细胞增殖和分化、促进肿瘤发生中发挥着至关重要的作用[12]。与抗心律失常药物胺碘酮类似,化合物WAG-003通过靶向KDM4A的TUDOR结构域中等抑制KDM4A的酶活性[13]。化合物N,N-desethylami也对KDM4A的TUDOR结构域具有抑制作用[18]。此外,共晶结构显示(PDB ID:5VAR)(见图1),化合物3可直接与TUDOR结构域的三甲基赖氨酸结合口袋结合而发挥抑制作用,为靶向KDM4酶家族的TUDOR结构域开发更高选择性和高效力的非催化功能抑制剂奠定基础[19]。
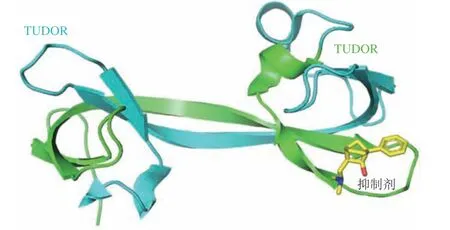
图1 化合物3靶向KDM4A非催化TUDOR结构域的晶体结构(PDB ID:5VAR)Figure 1 Crystal structure of compound 3 targeting the non-catalytic TUDOR domain of KDM4A (PDB ID: 5VAR)
1.2 靶向局部黏着斑激酶的非催化结构域研究现状
FAK也称为蛋白酪氨酸激酶2(protein tyrosine kinase 2,PTK2),是一种由PTK2基因编码的非受体型酪氨酸激酶,在整合素介导的信号转导通路中发挥着重要作用[20]。FAK由N端的4.1-埃兹蛋白-根蛋白-膜突蛋白(FERM)结构域、位于中心的激酶结构域和C端2个富含脯氨酸的基序即富含脯氨酸基序1(proline-rich 1,PR1)和PR2及黏着斑定位区FAT结构域组成(PDB ID:2QVX)[20]。其中位于中心的激酶结构域发挥酶催化功能。N端的FERM结构域介导FAK与整合素和生长因子受体等蛋白的直接相互作用,并通过与位于中心的激酶结构域的直接结合而阻止底物与催化结构域的结合,保护FAK免受肉瘤蛋白激酶(sarcoma,Src)磷酸化的激活。C端的FAT结构域包含多种蛋白-蛋白相互作用的结合位点,引导FAK到各种细胞的黏着斑复合物中[20-22]。Tyr397是FAK的一个关键位点,其自磷酸化介导了多个下游信号通路的激活[23]。在静息状态下,FERM结构域和中央激酶结构域形成自抑制分子内相互作用,FAK处于无活性状态。一旦FERM结构域和中央激酶结构域之间的分子内相互作用被阻断,FAK将被激活[20]。FAK的活化可增强Tyr397等位点的磷酸化,进而激活FAK依赖的信号通路,促进细胞的生长、增殖、生存和迁移等活动[24]。此外,FAK在转移性肿瘤中过表达,其激活介导的p53降解也在细胞的增殖和存活中发挥着关键作用,因而FAK是一个重要的肿瘤治疗靶点[20]。目前报道的FAK小分子抑制剂分为两大类:激酶依赖性酶功能抑制剂和激酶非依赖性支架功能抑制剂。其中激酶依赖性抑制剂如TAE-226、PF-573228、PF-562271、PF-4554878和GSK-2256098等均靶向FAK激酶结构域的腺嘌呤核苷三磷酸(ATP)结合位点,通过抑制FAK的Tyr397或Tyr861位点的磷酸化而抑制FAK的活性,但这类抑制剂在抑制细胞生长并诱导细胞凋亡方面具有一定的局限性[20,24-25]。而另一类激酶非依赖性小分子抑制剂如CFAK-C4通过靶向FAK非催化结构域的支架功能而发挥抑制作用[26-27]。血管生长因子受体3(vascular endothelial growth factor receptor 3,VEGFR-3)是一种受体酪氨酸激酶,参与肿瘤细胞及其附近的淋巴管内皮和淋巴结转移性肿瘤的淋巴管生成[28-29]。CFAK-C4是一种靶向FAK 的FAT结构域的VEGFR-3结合位点而开发的小分子抑制剂,可以破坏VEGFR-3与FAK的相互作用。临床前研究证明:CFAK-C4在包括胰腺癌、乳腺癌、神经母细胞瘤和晚期黑色素瘤等诸多癌症中呈现出治疗效果,目前已被美国FDA认定为孤儿药[23-24,30]。因此,靶向FAK非催化结构域的支架功能开发阻断FAK介导的肿瘤相关信号通路的高度特异性药物将成为一种可行且有潜力的抑癌策略[26]。
1.3 靶向蛋白质二硫键异构酶A1的非催化结构域研究现状
蛋白质二硫键异构酶A1(protein disulfide isomerase A1,PDIA1)是一种定位在内质网上的硫醇二硫醚氧化还原酶,可催化其底物蛋白上半胱氨酸残基之间二硫键的氧化、还原和异构化,是重要的折叠催化剂。PDIA1包含2种不同类型的类硫氧化还原结构域,即催化结构域(a和a')和非催化结构域(b和b')(PDB ID:4EKZ)(见图2)。2种类型结构域的序列同源性为37%,均具有Cys-Gly-His-Cys(CGHC)活性位点基序,但却独立地发挥二硫键氧化、还原和异构化功能。其中催化结构域a和a'发挥催化功能,非催化型结构域b和b'则通过支架功能发挥底物募集作用[31]。PDIA1参与如血小板活化、血栓形成和病毒感染等生物过程,其活性失调与诸多疾病如癌症、心血管和神经退行性疾病的发生发展密切相关。目前靶向PDIA1抑制剂可分为两大类:催化a型结构域抑制剂如RB-11-ca和KSC-34和非催化b型结构域抑制剂如ML359、Rutin和BAP2。其中,化合物ML359和Rutin具有一定的体外抑制血小板聚集的活性,可有效地阻断体内血栓的形成。并且这2个化合物均无明显的细胞毒性,为抗血栓的药物治疗奠定了基础[32-33]。化合物BAP2及其类似物通过靶向PDIA1的b型非催化结构域抑制DNA修复基因的表达进而达到抗肿瘤效果,因而BAP2可与DNA损伤剂联合使用从而联合治疗恶性胶质瘤[7,34]。
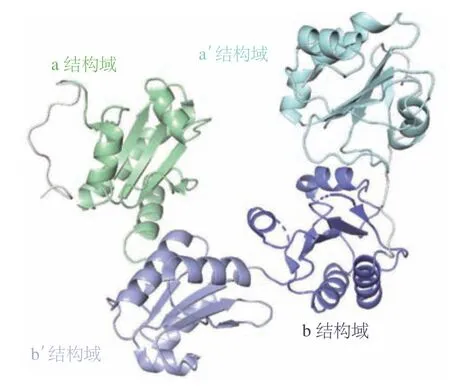
图2 蛋白质二硫键异构酶A1的晶体结构(PDB ID:4EKZ)Figure 2 Crystal structure of PDIA1 (PDB ID: 4EKZ)
2 变构调控策略
变构调节是指小分子化合物与酶蛋白分子活性位点之外的某一位点特异性结合,通过改变酶蛋白分子的构象变化而改变酶的活性[35]。与传统的作用于活性位点的药物相比,靶向变构位点的抑制剂具有更高的选择性、较低的脱靶毒性和较低的剂量要求等优势[35-36]。因而寻找调控蛋白酶变构调控机制的化合物也逐渐成为新药研发的新策略。目前已针对多个靶蛋白如含SH2结构域的蛋白酪氨酸磷酸酶2(src-homology 2 domain-containing protein tyrosine phosphatase-2,SHP2)、蛋白酪氨酸磷酸酶1B(protein tyrosine phosphatase 1B,PTP1B)、断裂点簇集区(breakpoint cluster region,BCR)-艾贝尔逊酪氨酸激酶(Abelson tyrosine kinase, ABL)和丝氨酸/苏氨酸蛋白激酶(serine/threonine kinase,AKT)等的变构调控机制成功开发了多个变构抑制剂[37-40],部分代表性的抑制剂如表2所示。
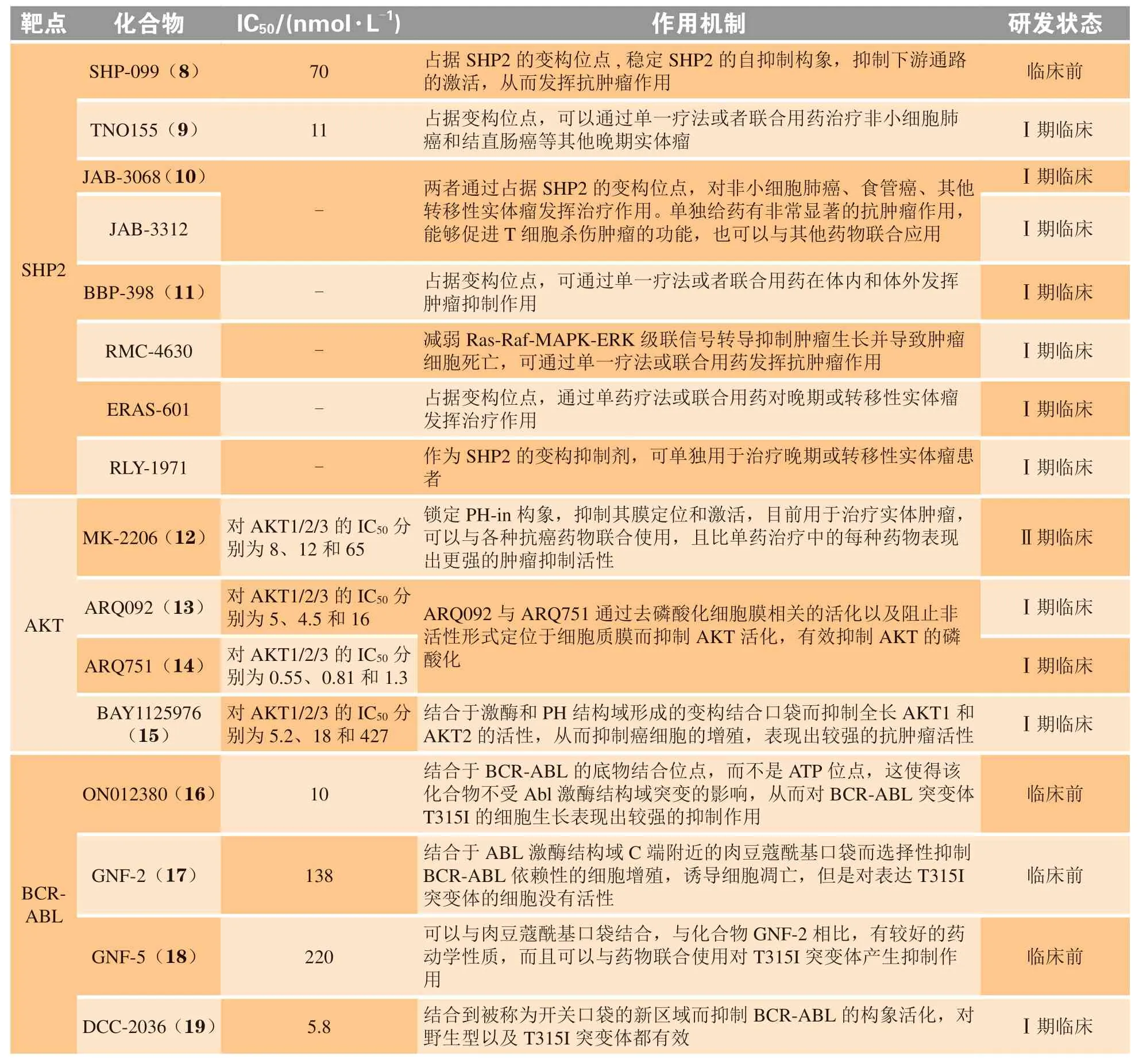
表2 变构调控策略相关靶点研究现状Table 2 Current status of research on targets with allosteric regulation strategy
2.1 靶向含SH2结构域的蛋白酪氨酸磷酸酶2的变构调控研究现状
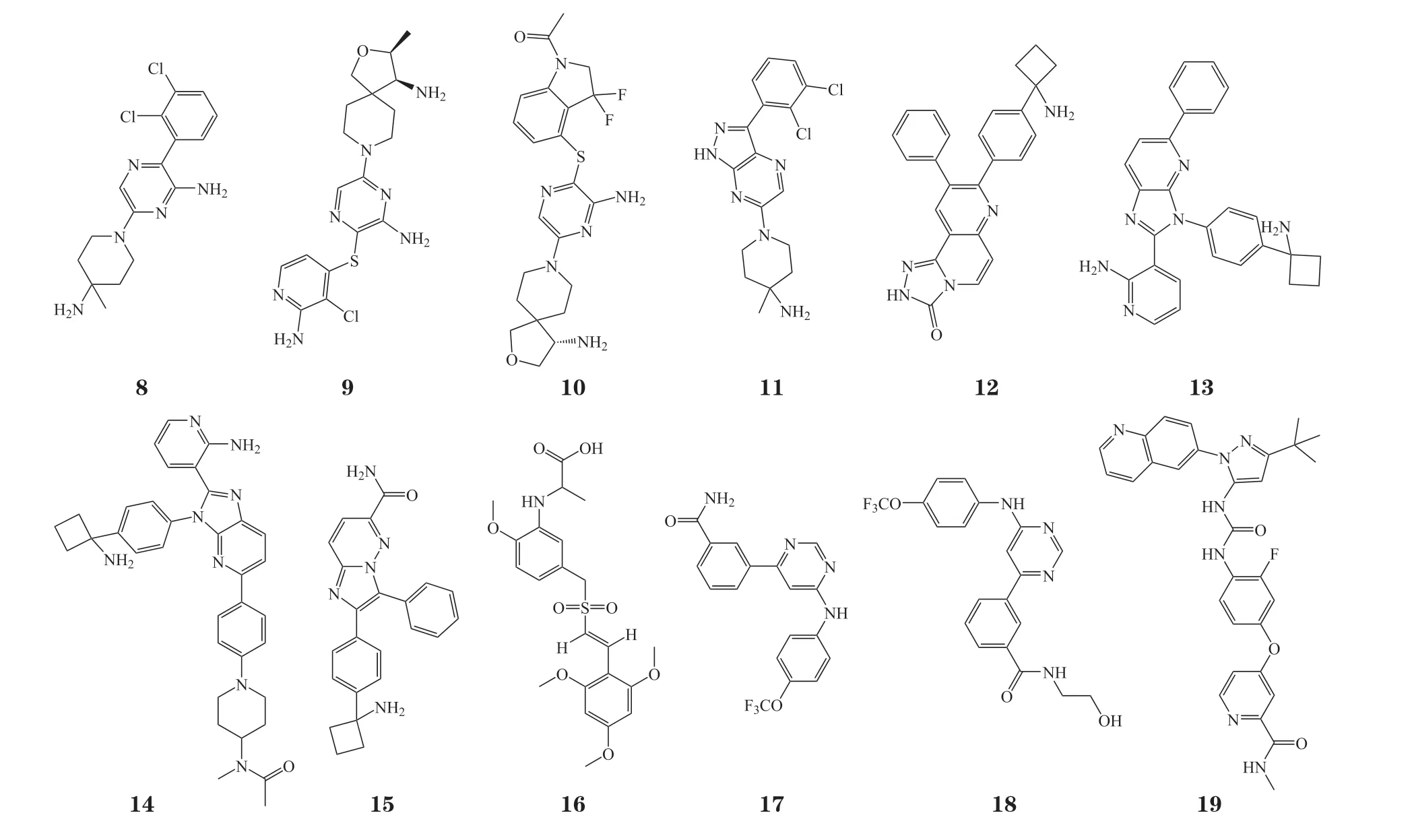
SHP2是由PTPN11基因编码的一种非受体酪氨酸磷酸酶,是多个信号通路如RAS-RAF-MAPK-ERK、磷脂酰肌醇-3-激酶(phosphatidylinositol 3-kinase,PI3K)-AKT-雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、Janus激 酶(Janus Kinase,JAK)-信号转导与转录激活因子(signal transducer and activator of transcription,STAT)等的关键调控因子[41-42]。SHP2是蛋白酪氨酸磷酸酶家族(protein tyrosine phosphatase,PTP)的一员,全长为593个氨基酸,由N末端的2个SH2结构域N-SH2和C-SH2,中间保守的蛋白酪氨酸磷酸酶催化结构域PTP和C端尾巴组成[43]。在基底状态下,SHP2采取自抑制构象(PBD ID:2SHP),N-SH2结构域被铆定到PTP结构域上,催化位点被堵塞。磷酸化蛋白的结合或激活突变将解除SHP2的自抑制状态,SHP2将采取开放构象(PBD ID:6CRF)(见图3)。N-SH2结构域将远离PTP结构域,催化位点被释放,SHP2的酶活性被激活,进而激活下游的级联信号通路[44-45]。临床研究表明,SHP2的激活或过表达与努南综合征、青少年单核粒细胞白血病、豹皮综合征等多种类型疾病的发生密切相关,是治疗疾病的潜在药物作用靶点[46-48]。目前已经报道了多个靶向SHP2催化位点的中等活力小分子抑制剂,如NSC-87877、Ⅱ-B08和cefsulodin衍生物等,这些抑制剂可通过下调SHP2的PTP活性而达到治疗疾病的目的。通过对多样性小分子化合物库的筛选,笔者所在课题组也发现了2种结构新颖的SHP2的PTP抑制剂SYK-85和WS-635[49]。但由于PTP家族催化结构域的序列保守性,这些药物的特异性通常较低,并且PTP催化位点的高度溶剂化和极性性质也阻碍了SHP2小分子抑制剂的临床开发[50-60]。为了克服以上缺点,2016年诺华团队基于SHP2独特的变构调控机制建立了一套多重高通量筛选方案,开发了首个高效、高选择性且具有良好口服生物利用度的SHP2变构抑制剂SHP-099,SHP-099的开发为后续SHP2变构抑制剂的开发提供了结构骨架[44]。截至目前,已有8个高效、高选择性且口服有效的变构SHP2小分子抑制剂,即TNO155、RMC-4630、RLY-1971、JAB-3068、JAB-3312、BBP-398、ERAS-601和SH3809进入临床研究,以评估其单用或联用在实体瘤和获得性耐药癌症中的治疗效果[61-63]。作为 “分子胶水”,这些化合物通过结合在N-SH2、C-SH2和PTP的相互作用界面上稳定SHP2的自抑制构象,变构地抑制SHP2的酶活性。在体外可通过调控SHP2介导的相关信号通路而抑制癌细胞的增殖,进而达到治疗癌症的效果[44,64]。临床前数据表明,TNO155与表皮生长因子受体(epidermal growth factor receptor,EGFR)、小鼠肉瘤病毒癌基因同源体B(murine sarcoma viral oncogene homolog B,BRAF)、Kirsten大鼠肉瘤病毒癌基因G12C突变(Kirsten rat sarcoma viral oncogene G12C mutation,KRASG12C)和细胞周期蛋白依赖性激酶(cyclin-dependent kinase,CDK)4/6抑制剂以及程序性死亡受体1(programmed death-1,PD-1)抗体联合使用均呈现出较好的协同疗效[65]。自2007年以来,单一TNO155或TNO155与PD-1抗体spartalizumab、CDK4/6抑制剂ribociclib、BRAF抑制剂dabrafenib (Tafinlar®)、口服ERK抑制剂LTT462或KRASG12C特异性口服抑制剂如MRTX849、JDQ443和JDQ443联合使用已经进入了Ⅰ/Ⅱ期临床研究,以确定最大耐受剂量和推荐剂量下药物的抗肿瘤活性、安全性、耐受性和PK/PD性质,为获得性耐药患者提供了另一种治疗策略。药理学研究表明,在osimertinib耐药的非小细胞肺癌(nonsmall cell lung cancer, NSCLC)模型中,BBP-398单独或与EGFR抑制剂osimertinib联合使用在体内外均呈现出有效的肿瘤抑制作用,且可恢复NSCLC对osimertinib的敏感性[66]。RMC-4630可通过干扰RAS-RAF-MAPK-ERK级联信号而抑制肿瘤生长,最终导致肿瘤细胞死亡。单用RMC-4630或与MEK抑制剂cobimetinib协同使用均呈现较好的抗肿瘤作用。此外,RMC-4630可与KRASG12C抑制剂AMG510联合使用治疗KRASG12C突变的晚期实体肿瘤患者。ERAS-601和RLY-1971单用或与其他药物联合使用也对晚期或转移性实体瘤发挥治疗作用。此外,由北京加科思新药研发有限公司自主开发的变构SHP2抑制剂JAB-3068和JAB-3312也已获批开展针对NSCLC、头颈部癌、食管癌和其他转移性实体瘤患者的安全性、耐受性、药动学和初步抗肿瘤活性的临床研究[42]。
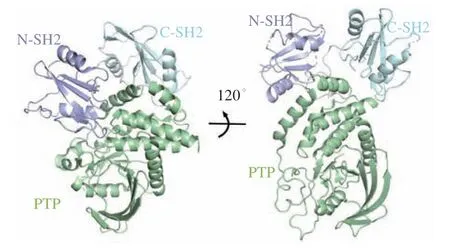
图3 SHP2的自抑制构象(PBD ID:2SHP)与开放构象(PBD ID:6CRF)Figure 3 Auto-inhibited (PBD ID: 2SHP) and open conformation of SHP2 (PBD ID: 6CRF)
2.2 靶向丝氨酸/苏氨酸蛋白激酶的变构调控研究现状
AKT也被称为蛋白激酶B(protein kinase B,PKB),是PI3K信号通路的重要成员,其过表达或激活与乳腺癌、结肠癌和卵巢癌等一系列癌症的发生和发展密切相关,是癌症治疗的一个潜在药物作用靶点[67-69]。AKT家族包括AKT1 ~ 3这3种亚型,均由N末端的PH结构域、中心的激酶结构域(kinase domain,KD)和C末端的调节结构域3部分组成(PDB ID:6HHG)[70](见图4)。3种亚型的激酶结构域高度保守,同源性超过85%。但它们的PH结构域在不同的亚型中存在较大的差异,同源性约为60%,可促进AKT与膜磷脂的黏附[68]。AKT的N末端PH结构域与中心的KD结构域之间的分子内相互作用介导了AKT的“PH-in”和“PH-out”2种构象的转换。PH与KD结构域间的分子内相互作用可使AKT处于非活化的“PH-in”构象,防止AKT激酶结构域的活性环被磷酸肌醇依赖性蛋白激酶-1(PI3K-dependent kinase 1,PDK1)磷酸化,但当二者之间的相互作用受到破坏后,AKT转变为“PHout”构象,激酶结构域的活性环可被PDK1磷酸化,进而导致AKT的活化[36,68]。由于AKT激酶家族中ATP结合位点的保守性,因而靶向AKT激酶结构域的ATP竞争抑制剂面临着选择性和脱靶副作用的挑战,多项临床试验已终止[37]。因而针对AKT的变构调控机制,开发高选择性和低毒性的ATP非竞争性变构抑制剂逐渐引起了研究者的关注[68,71]。目前报道的处于临床试验阶段的AKT变构抑制剂主要有MK-2206、ARQ092、ARQ751和BAY1125976[72]。MK-2206是一种高效、高选择性的新型非ATP竞争性变构AKT抑制剂[73]。单用MK-2206或MK-2206与细胞毒性药物如拓扑异构酶抑制剂(阿霉素和喜树碱)、抗代谢物(吉西他滨和5-氟尿嘧啶)、抗微管药物(多西紫杉醇)和DNA交联剂(卡铂)联用均在体内和体外呈现出抗肿瘤活性,并且耐受性良好[74]。临床研究表明,虽然MK-2206的半衰期较长,可连续隔日服用,但仍具有皮疹、疲劳和胃肠道毒性等缺陷,因此仍需针对该化合物进行进一步的优化[75]。ARQ092是一种可口服、高效且高选择性的变构AKT抑制剂,在实体瘤和血液肿瘤中具有抗肿瘤活性,但仍具有轻度至中度的高血糖、皮疹、腹泻、呕吐、黏膜炎和转氨酶升高等药物相关不良反应[76]。与ARQ092相比,ARQ751是一种具有更高效力和选择性的AKT变构抑制剂[77]。生化和细胞分析表明,这2种化合物均可有效地调控AKT介导的相关信号通路从而抑制AKT的活性,进而抑制诸多肿瘤细胞的增殖,其中在白血病、乳腺癌、子宫内膜癌和结直肠癌细胞系中效果最强[77]。BAY1125976也是一种有效的高选择性变构AKT抑制剂,通过结合到由PH和KD结构域形成的变构结合口袋而选择性地抑制AKT1和AKT2的活性。BAY1125976呈现较好的疗效和体内耐受性,并在多个PI3K/AKT/mTOR通路激活的肿瘤模型中显示出剂量依赖性的抗肿瘤疗效[78]。相比于MK-2206,BAY1125976的半衰期较短,在临床用药过程中也有高血糖、皮疹等现象出现,因而也仍需进一步的优化[79]。
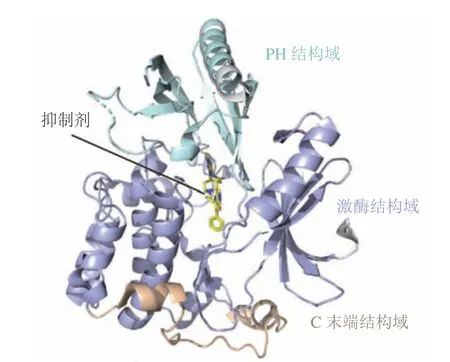
图4 靶向AKT变构位点的抑制剂共晶结构(PDB ID:6HHG)Figure 4 Co-crystal structure of inhibitors targeting the allosteric site of AKT (PDB ID: 6HHG)
2.3 靶向断裂点簇集区-艾贝尔逊酪氨酸激酶的变构调控研究现状
BCR-ABL是由断点聚集区蛋白BCR与ABL蛋白融合而成的一种相对分子质量约为210 000的非受体型酪氨酸蛋白激酶。BCR-ABL可催化γ-磷酸基团从ATP转移到蛋白中特定酪氨酸残基的羟基 上,参 与RAS-RAF-MAPK-ERK、PI3K-AKT、JAK-STAT和髓细胞组织增生蛋白(myelocytomatosis proteins,Myc)等多种信号通路[39,80]。ABL蛋白由N端无序的Cap结构域、SRC同源结构域3(Src homology 2,SH3)、SH2、C端激酶结构域KD、SH3与SH2连接区(ContectorSH3/2)和SH2与KD连接区(LinkerSH2/KD)构成。SH2、SH3和KD 3个结构域之间的分子内相互作用可变构地调控ABL蛋白的酶活性。N端的肉豆蔻酰基插入C端的口袋后将稳定ABL的KD结构域,使ABL呈现自组装的抑制构象。当SH2结合到N端的KD结构域,ABL呈现扩展的激活构象[23,81]。单独的ABL蛋白可在细胞核和细胞质之间穿梭,但当其与BCR蛋白融合后,ABL蛋白将失去穿梭特性,停留在细胞质内。BCR与ABL蛋白的融合可激活ABL的酪氨酸激酶活性,通过与致癌途径中蛋白质的相互作用而导致癌症的发生[82]。临床研究发现BCR-ABL融合蛋白与慢性粒细胞白血病的发生密切相关[39]。目前报道的靶向BCR-ABL抑制剂分为两类: 1)ATP竞争性抑制剂如imatinib、nilotinib和PPY-A等,可与活化或非活化构象的BCR-ABL蛋白相结合; 2)ATP非竞争性变构抑制剂如ON012380、GNF-2及其衍生物GNF-5和DCC-2036等,可结合到酶的底物结合位点或远离ATP结合位点的变构口袋中。但ATP竞争性抑制剂却面临着耐药性的挑战,而单用ATP非竞争性变构抑制剂或与ATP竞争抑制剂联合使用可在一定程度上克服单一药物的耐受突变[83-85]。ON012380可与BCR-ABL的底物结合位点而非ATP结合位点相结合,因而这些化合物不受ABL激酶突变的影响,对BCR-ABL突变体T315I细胞的生长也呈现出较好抑制效果[86-87]。GNF-2及其衍生物GNF-5是一类高选择性的变构BCR-ABL抑制剂,通过与ABL酶C末端的豆蔻酸盐结合口袋相结合,诱导稳定BCR-ABL的非活化构象[88]。相比于GNF-2,GNF-5具有更好的药动学特性。研究表明,GNF-5可与ATP竞争性抑制剂imatinib联合使用而改变ATP结合位点的构象,协同抑制耐药突变[89]。DCC-2036是一种全新的酪氨酸激酶抑制剂,通过结合到一个称为“开关口袋”的新区域而抑制BCR-ABL的构象活化[90]。研究显示,DCC-2036也表现出良好的生物利用度和安全性,在imatinib耐药性慢性粒细胞白血病治疗方面具有较大的潜力[83]。
3 蛋白-蛋白相互作用策略
蛋白质和蛋白质之间的相互作用在诸多重要的细胞功能中发挥着至关重要的作用,对于生物体维持正常的生理功能是必不可少的。除此之外,蛋白-蛋白相互作用也与众多疾病如癌症和自身免疫性疾病的发生密切相关[91-92]。如胚胎外胚层发育蛋白(embryonic ectoderm development,EED)与Zeste基因增强子同源物2(enhancer of zeste homolog 2,EZH2)之间的相互作用可促进癌细胞的增殖,WD40重复蛋白5(WD40 repeat protein 5,WDR5)与混合谱系白血病1蛋白(mixed lineage leukemia 1,MLL1)之间相互作用与急性白血病的发生密切相关[93-94]。因此,开发靶向蛋白-蛋白相互作用界面的抑制剂在疾病研究领域至关重要。但是与传统的单一靶点相比,蛋白-蛋白相互作用界面具有大而平坦且疏水的特点,是高选择性小分子抑制剂的发现和开发过程中需要面临的问题[95]。目前已报道了多个成功靶向蛋白-蛋白相互作用界面如EED-EZH2、MLL-WDR5、鼠双微粒体2(murine double minute 2,MDM2)-p53、B细胞淋巴瘤(B cell lymphoma,BCL-2)-B细胞淋巴瘤相关X蛋白(B cell lymphoma associated X protein,Bax)、表面抗原分化簇4(cluster of differentiation 4,CD4)-糖蛋白120(glycoprotein 120,gp120)、RAS-RAF和Kelch 样环氧氯丙烷相关蛋白1(Kelch-like ECHassociated protein 1,KEAP1)-核因子E2相关因子2(nuclear factor erythroid-2-related factor 2,NRF2)等开发抑制剂的案例,部分代表性抑制剂如表3所示。
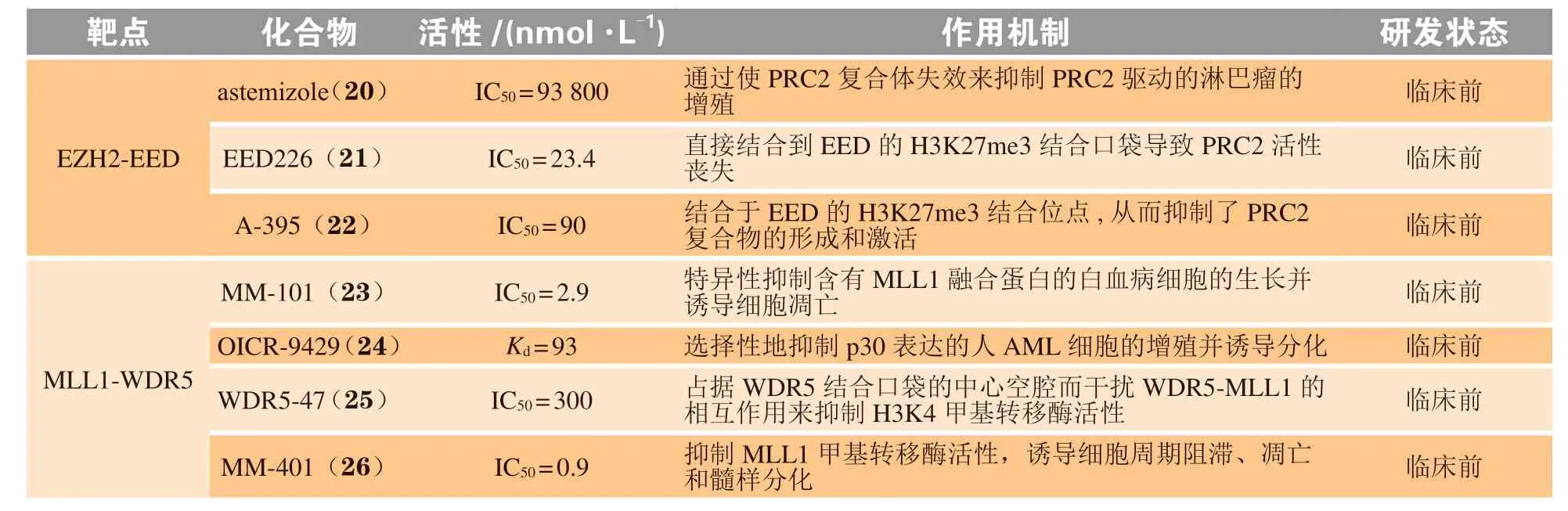
表3 蛋白-蛋白相互作用策略相关靶点研究现状Table 3 Current status of research on targets with protein-protein interaction strategy
3.1 靶向胚胎外胚层发育蛋白与Zeste基因增强子同源物2的相互作用研究现状
多梳抑制复合体2(polycomb repressive complex 2,PRC2)由3个核心亚基EZH2、EED和Zeste 12基因抑制因子(suppressor of zeste 12,SUZ12)组成,在转录调控、细胞周期、衰老和细胞分化等过程中发挥着关键作用[96]。通过催化亚基EZH2和EED之间的蛋白-蛋白相互作用,PRC2可通过调控H3K27me3的甲基化调节染色质的结构和转录抑制[97]。PRC2的异常活化与癌症的发生发展密切相关,是潜在的表观遗传癌症治疗靶点[98]。抑制EZH2和EED之间的相互作用可使PRC2复合物失去活力,通过抑制癌细胞的增殖而达到治疗癌症的目的[93]。近几年报道的靶向EZH2-EED之间相互作用界面的高效、高选择性的小分子抑制剂主要有astemizole、EED226和A-395[96-97,99]。研 究 表 明,astemizole可通过干扰EZH2和EED之间的相互作用而破坏PRC2复合物的稳定性,进而影响其甲基转移酶活性而抑制PRC2驱动的淋巴瘤细胞的增殖[97]。结构研究表明,抑制剂A-395和EED226通过直接结合到EED的H3K27me3结合口袋而阻止PRC2的活化[96,99]。这些抑制剂的发现为进一步开发更高效且高选择性的PRC2抑制剂奠定了基础。
3.2 靶向混合谱系白血病1蛋白与WD40重复蛋白5的相互作用研究现状
MLL1是一种组蛋白H3K4甲基转移酶,WDR5是激活MLL1酶活性所必需的多蛋白复合物的重要组成部分。MLL1与WDR5的相互作用在急性淋巴细胞系白血病(acute lymphoid leukemia,ALL)和AML的发展中发挥着重要作用,因此靶向MLL1蛋白或MLL1与WDR5蛋白-蛋白相互作用界面开发抑制剂为急性白血病的治疗提供了机会[100]。近几年报道的作用于MLL1-WDR5蛋白-蛋白相互作用界面的抑制剂主要有拟肽类抑制剂如MM-101及其衍生物和小分子抑制剂如OICR-9429和WDR5-47等[101-102]。拟肽类抑制剂MM-101和MM-401可通过模拟MLL1与WDR5的结合模式而抑制MLL1的H3K4甲基转移酶活性,为进一步开发更有效的混合谱系白血病药物奠定了基础[100,103]。高亲和力和高选择性小分子抑制剂OICR-9429可通过与WDR5直接结合,竞争性地破坏MLL与WDR5之间的相互作用。由于WDR5参与p30诱导的自我更新和白血病发生,OICR-9429可选择性地抑制p30表达的人白血病细胞的增殖和分化,因此其对白血病的治疗发挥着重要作用[104]。此外,相对分子质量较低的小分子拮抗剂WDR5-47也对WDR5具有中等的亲和力,可通过占据WDR5结合口袋的中心空腔而强效地抑制MLL1与WDR5之间的相互作用,该化合物的发现为进一步设计优化较高亲和力的化合物提供了结构骨架[105]。
4 结语与展望
近年来,靶向蛋白的非催化功能开发抑制剂受到广泛关注,诸多成功案例已被报道,为该类抑制剂在相关疾病领域的临床应用奠定了基础。蛋白非催化结构域策略通过靶向蛋白的非催化结构域开发抑制剂来达到治疗效果;变构调控策略是通过靶向蛋白的变构位点进行新药研发;蛋白-蛋白相互作用策略则是通过靶向蛋白-蛋白相互作用界面开发高选择性的小分子抑制剂。3种靶向蛋白非催化功能的药物开发新策略在药物研究领域均具有广阔的前景,但仍存在不足之处。目前针对蛋白非催化结构域的药物开发仍在临床前初期研究阶段,缺乏深入的药物化学、活性机制、毒性和成药性等临床前评价研究。相比于活性位点抑制剂,针对变构位点的抑制剂具有更高的选择性以及较低的毒性、脱靶毒性和更低的剂量要求[106-107]。但是由于变构位点相比正构位点具有更低的进化压力,变构位点发生耐药突变的机会增加,变构药物面临着耐药性的挑战。因此研究变构调节策略中的耐药突变机制对于解决这一挑战具有重要的意义[107]。由于蛋白质-蛋白质界面大且平坦,获得高活性靶向蛋白相互作用的小分子抑制剂在新药研发领域仍具有一定的挑战[108]。此外,靶蛋白非催化功能的作用机制及其靶向开发策略等领域的研究仍相对较少,在一定程度上限制了非催化功能抑制剂的开发。因此,开发针对靶蛋白非催化功能的抑制剂设计策略、深入探究该类抑制剂的作用机制等是该领域重要的研究方向。总的来说,非催化功能抑制剂在疾病领域具有非常大的潜力,靶向非催化功能的抑制剂的开发是药物研发领域的重要研究方向,有望在未来几年取得突破进展。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
汽车工程师(2021年12期)2022-01-18
第一财经(2019年8期)2019-08-26
分析化学(2018年4期)2018-11-02
科学24小时(2018年1期)2018-01-10
作文·初中版(2017年6期)2017-06-16
中文信息(2017年2期)2017-04-13
江苏农业科学(2016年11期)2017-03-21
现代养生·下半月(2016年6期)2016-10-21
智能计算机与应用(2016年4期)2016-09-26
