
高效液相色谱-同位素稀释-串联质谱法测定人源瘦素的含量
2015-12-14 08:23孙雪晴胡高飞宋德伟田亚平颜光涛李红梅
质谱学报 2015年1期
孙雪晴,胡高飞,宋德伟,田亚平,颜光涛,徐 蓓,李红梅
(1.北京化工大学,北京 100029;2.中国计量科学研究院,北京 100013;3.解放军总医院生化科,北京 100853)
高效液相色谱-同位素稀释-串联质谱法测定人源瘦素的含量
孙雪晴1,2,胡高飞1,宋德伟2,田亚平3,颜光涛3,徐 蓓2,李红梅2
(1.北京化工大学,北京 100029;2.中国计量科学研究院,北京 100013;3.解放军总医院生化科,北京 100853)
为准确测定人源瘦素的含量,建立了蛋白水解-高效液相色谱-同位素稀释-串联质谱(HPLCIDMS)测定人源瘦素纯品绝对含量的方法,并对人源瘦素蛋白水解时间进行优化。结果表明,人源瘦素中的被测氨基酸在温度为110℃,6mol/L HCl溶液中水解40h可达平衡。水解样品通过高效液相色谱分离后,使用多反应监测模式(MRM)分别检测脯氨酸m/z 116>m/z 70(Pro)和m/z 121>m/z 74(标记Pro)离子对,缬氨酸m/z 118>m/z 72(Val)和m/z 123>m/z 76(标记Val)离子对,亮氨酸m/z132>m/z86(Leu)和m/z142>m/z96(标记Leu)离子对,根据检测结果计算得出人源瘦素纯品的含量为0.582g/g,扩展不确定度为0.014g/g(k=2),这与纯度扣除法测定结果基本一致,标准偏差为4.8%。该方法提高了人源瘦素含量测定的准确度和精密度,使测量结果可溯源至SI单位,为人源瘦素标准物质的研制奠定了基础。
人源瘦素;准确定量;同位素稀释质谱法;标准物质;溯源性
人源瘦素(leptin,LP)是一种由肥胖基因编码的蛋白质类激素[1-2],其原型包含167个氨基酸残基,在分泌人血的过程中,去除21个N-末端信号肽中的残基,形成含有146个氨基酸残基的分泌型成熟瘦素,其相对分子质量为16 ku[3-5],无糖基化[4]。人源瘦素主要产生于脂肪组织,并在胎盘、卵巢、乳腺上皮细胞、骨髓和淋巴组织中表达[6-8],至少有6种受体,这些受体在脂肪细胞、肾上腺皮质细胞、睾丸间质细胞、脉络丛、下丘脑和骨骼肌细胞中均有表达[9]。人源瘦素通过与其特异性受体结合,可在中枢神经系统和外周组织中发挥生物学效应,在涉及肥胖、食欲、食物摄取、能量消耗和维持机体能量平衡中起重要作用[10]。自发现瘦素以来,科研工作者做了大量的基础和临床研究,发现瘦素在肺癌[11]、结肠癌[12]、乳腺癌[13]等恶性肿瘤及睡眠障碍[3]中高表达,有诱激肿瘤细胞增殖,促进肿瘤血管生成及肿瘤侵袭、转移等作用[14]。
目前,人源瘦素在许多疾病中的作用及机理研究仍处于起步阶段,临床常用酶联免疫法(enzyme-linked immunosorbent assay,ELISA)体外检验人源瘦素[10-11],但此方法耗时长、费用高、操作复杂,且不同厂家提供的试剂盒检测结果不具有跨时空的可比性。因此,亟需建立人源瘦素的参考测量方法,并研制出相应的标准物质,以使人源瘦素的标准化检测得以实现。
同位素稀释质谱法是目前国际公认的测量微量、痕量和超痕量元素的权威方法[15]。将已知质量和丰度的稳定同位素作为稀释剂加入样品中,混合均匀后用质谱测定混合前后同位素丰度的变化,由此可计算出样品中该元素的含量。目前,此方法已应用于前列腺特异抗原[16-17]、C-反应蛋白等定量工作中,并获得了与酶联免疫法一致的检测结果,线性相关系数R2=0.97,且检测限可降低10倍[18-19]。
本工作拟建立同位素稀释质谱法定量测定人源瘦素的绝对含量,使检测结果可溯源至SI单位制,为建立人源瘦素的标准定值体系及研制相关标准物质奠定基础。
1 实验部分
1.1 主要仪器与装置
Agilent 6410型高效液相色谱-串联质谱联用仪:美国Agilent公司产品;XP26型分析天平(最大量程22g,精度0.001mg):瑞士Mettler-Toledo公司产品;ME235S型分析天平(最大量程230g,精度0.01mg):德国Sartorius公司产品;UFE500型烘箱:德国Memmert公司产品;Vacucell型真空干燥箱:德国MMM公司产品;N-EVAP111型氮吹仪:美国Organomation公司产品;涡旋震荡仪:美国Scilogex公司产品;垂直电泳槽:美国伯乐公司产品;KS260型摇床:德国IKA公司产品;Milli-Q型超纯水系统:美国Millipore公司产品;基质辅助激光解吸电离飞行时间质谱仪(MALDI-TOF-MS):德国Bruker公司产品。
1.2 主要材料与试剂
人源瘦素(纯度98%):美国Pepro Tech公司产品;乙腈(色谱纯):美国J.T Baker公司产品;三氟乙酸(色谱纯)、全氟庚酸:美国Sigma公司产品;甲酸(分析纯)、盐酸(优级纯)、冰醋酸(优级纯):北京化学试剂公司产品;缬氨酸(Val,纯度99.0%,GBW(E)100055)、脯氨酸(Pro纯度99.0%,GBW(E)100084)、亮氨酸(Leu纯度99.0%,GBW(E)100058):由中国计量科学研究院提供;13C5-L-缬氨酸、13C5-L-
脯氨酸、13D10-L-亮氨酸(纯度98%):美国CIL公司产品;蛋白预制胶:美国伯乐公司产品;Tris base(优级纯):美国Angus公司产品;甘氨酸(优级纯):中国Solarbio公司产品;十二烷基磺酸钠(Sodium dodecyl sulfate,SDS):北京鼎国昌盛生物技术有限责任公司产品;蛋白银染试剂盒:中国康为世纪公司产品;彩虹预染宽分子质量蛋白Marker(10-260KD):中国中科泰瑞公司产品。
1.3 聚丙烯酰胺凝胶电泳(SDS-PAGE)实验
将蛋白质预制胶放入垂直电泳槽内,倒入电泳缓冲液,充分浸泡预制胶后,将人源瘦素样品和上样缓冲液以1∶1的比例加入1.5mL离心管中,混匀,离心,然后在100℃水浴锅中煮10min,取出后离心;将彩虹预染宽分子质量蛋白Marker从-20℃取出,放至室温,离心待用。
加入人源瘦素样品和蛋白Marker后,设置电泳电压100V,时间50min。电泳完成后,按照蛋白银染试剂盒说明书进行银染。
1.4 MALDI-TOF-MS检测人源瘦素的相对分子质量
配制基质溶液1:芥子酸(sinapic acid,SA)在乙醇中的饱和溶液,离心后取上清液;配制基质溶液2:SA溶于TA30(30%乙腈溶于0.1% TFA)的饱和溶液,离心后取上清液。取0.5μL基质溶液1点板,自然晾干后,取1μL样品与基质的等比混合液点板,待测。
1.5 同位素稀释质谱法定量测定人源瘦素
1.5.1 样品及标准品的配制 1)精确称取0.49mg人源瘦素于958.29mg水中,配制成浓度为0.511 1mg/g的人源瘦素储备液;2)分别配制脯氨酸、缬氨酸、亮氨酸混合储备液及其同位素标记的氨基酸混合储备液;3)取25μL人源瘦素储备液并称重,加入一定量标记的混合氨基酸储备液,使之与人源瘦素水解后的氨基酸含量比值约为1;4)配制低标溶液:分别量取非标记的氨基酸和标记的氨基酸混标溶液并称重,使两者含量比值约为0.8;5)配制高标溶液:分别量取非标记的氨基酸和标记的氨基酸混标溶液并称重,使两者含量比值约为1.2。
1.5.2 样品水解条件 将准确称量的1.5.1节中的样品(3)置于40℃真空干燥箱中烘干,待样品冷却至室温后,加入500μL 6mol/L盐酸,通氮除氧,密封,置于(110±0.5)℃烘箱中水解40h;水解后的样品用氮吹仪吹干,用1 000μL 0.1%甲酸水溶液复溶;经0.22μm滤膜过滤后,用液相色谱-质谱仪测定。
1.5.3 色谱条件 色谱柱:Agilent SB-Aq C18柱(150mm×2.1mm×5μm);进样量8μL;柱温40℃;流动相:0.8mmol/L全氟庚酸和0.05%三氟乙酸的水溶液(A),乙腈(B);流动相梯度列于表1。
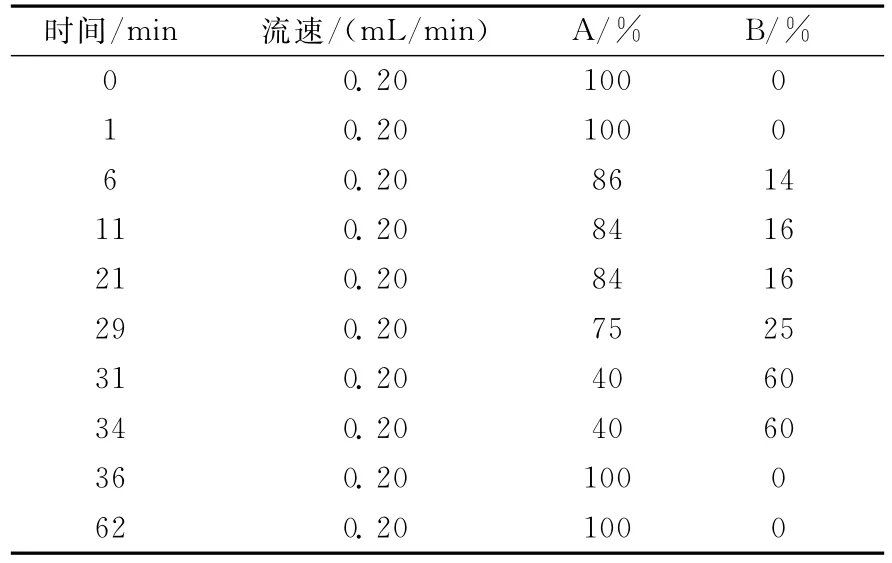
表1 定量测定人源瘦素的流动相梯度Table 1 The gradient solutions for human leptin quantitation
1.5.4 质谱条件 电喷雾电离源(ESI),正离子扫描模式,干燥气流速8L/min,毛细管温度350℃,毛细管电压4 000V,多离子监测模式(MRM)。对于脯氨酸,分别监测m/z 116>m/z70(Pro)和m/z121>m/z74(标记Pro)离子对;对于缬氨酸,分别监测m/z 118>m/z 72(Val)和m/z123>m/z 76(标记Val)离子对;对于亮氨酸,分别监测m/z132>m/z86(Leu)和m/z142>m/z 96(标记Leu)离子对,通过同位素稀释质谱法进行定量测定。
1.5.5 人源瘦素含量的计算 采用同位素稀释质谱法测定人源瘦素的含量,氨基酸的浓度ca(μg/g)按式(1)进行计算:

式中:P为氨基酸标准物质的纯度;PH为人源瘦素的水解效率(实验默认人源瘦素在最优化条件下完全水解);m标为所加标记氨基酸的质量(g);R样为样品中氨基酸和同位素标记氨基酸的峰面积比;I1为低标溶液中氨基酸和同位素标记氨基酸的质量比;I2为高标溶液中氨基
酸和同位素标记氨基酸的质量比;R1为低标溶液中氨基酸和同位素标记氨基酸的峰面积比;R2为高标溶液中氨基酸和同位素标记氨基酸的峰面积比;M为所加样品质量(g)。
将ca代入式(2),计算人源瘦素的浓度cLP(μg/g):

式中:NLP为人源瘦素的分子质量;n为人源瘦素蛋白序列中对应的氨基酸个数(其中,脯氨酸6个,缬氨酸11个,亮氨酸23个);Na为氨基酸的分子质量。
按照式(3)计算人源瘦素的质量分数A:

式中:A为人源瘦素蛋白的质量分数(g/g);mLP为称取人源瘦素蛋白的质量(g);m水为溶解人源瘦素蛋白的水的质量(g)。
2 结果与讨论
2.1 人源瘦素的聚丙烯酰胺凝胶电泳结果
人源瘦素的聚丙烯酰胺凝胶电泳结果示于图1。可以看出,在样品点样通道上除了人源瘦素条带外并无其他明显的条带出现,表明样品中人源瘦素蛋白的纯度较高,无其他杂质蛋白质存在,因此,利用蛋白水解-同位素稀释质谱法进行定量检测的结果较为可信。由于蛋白形态的不同,会导致其在聚丙烯酰胺凝胶电泳中的迁移速率不同,因此,采用聚丙烯酰胺凝胶电泳检测的人源瘦素相对分子质量的结果会有偏差。
2.2 MALDI-TOF-MS检测人源瘦素样品相对分子质量的结果
通过MALDI-TOF-MS对人源瘦素样品进行检测,得到样品的分子质量为16 160u,示于图2。该结果与人源瘦素的理论相对分子质量一致,这为下一步的同位素稀释-液相色谱-串联质谱定量检测人源瘦素样品的计算奠定了基础。
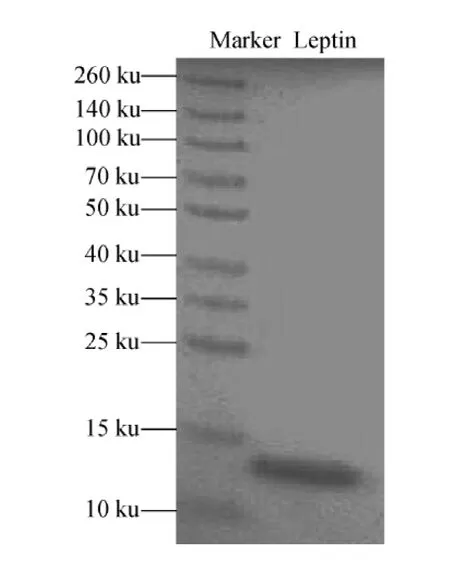
图1 人源瘦素的聚丙烯酰胺凝胶电泳图Fig.1 SDS-PAGE of the human leptin

图2 MALDI-TOF-MS检测人源瘦素分子质量的质谱图Fig.2 Mass spectrogram of the human leptin detected by MALDI-TOF-MS
2.3 人源瘦素水解后的氨基酸的高效液相色谱分离结果
采用MRM监测人源瘦素水解后的脯氨酸、缬氨酸和亮氨酸离子对,3种氨基酸可被高效液相色谱完全分离,谱图示于图3。
2.4 人源瘦素的水解时间
水解时间过长或者过短都会导致样品水解不完全,从而影响定量结果的准确性。对9个平行样品检测不同水解时间的氨基酸含量,结果示于图4。可以看出,水解时间为40h时,人源瘦素蛋白水解达到平衡,进一步水解会导致氨基酸含量的略微降低。因此,确定水解时间为40h。
2.5 同位素稀释质谱法测定人源瘦素的含量
分别测定6个平行样品,根据检测结果,按式(4)分别由3种氨基酸浓度计算人源瘦素蛋白的含量,结果列于表2。

由表2可知,6个样品中由脯氨酸、缬氨酸和亮氨酸计算得到的人源瘦素质量分数的平均值分别为0.638、0.531、0.577g/g,取三者的平均值得到高效液相色谱-同位素稀释-串联质谱法检测人源瘦素的质量分数为0.582g/g,
6个样品测定结果的RSD为5.2%,说明此方法检验人源瘦素的重复性较好。

图4 人源瘦素水解效率随时间的变化Fig.4 The hydrolysis efficiency of human leptin changes with time
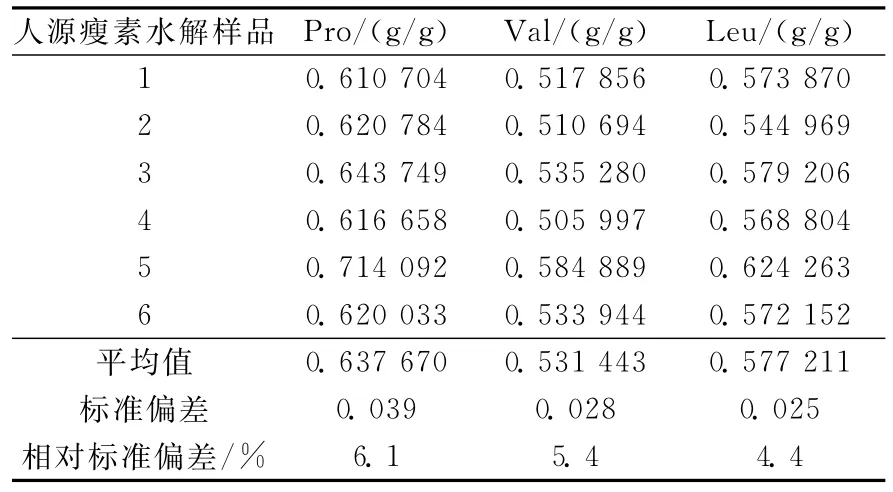
表2 由同位素稀释质谱法检测所得的氨基酸浓度计算人源瘦素蛋白的质量分数Table 2 Human leptin mass fraction calculated by the concentration of amino acids determined through IDMS
采用高效液相色谱法检测人源瘦素样品的纯度为94.01%,其中包括水分、灰分等无紫外吸收的成分,因此取6个样品于真空干燥箱中,每隔4h称重至恒重,计算得出样品中的水分为28%;使用ICP-MS法对样品中的无机元素进行半定量检测,得出人源瘦素样品中的无机元素含量为15.8%;利用纯度扣除法按式(4)计算得出人源瘦素的质量分数为52.8%。由同位素稀释-液相色谱-串联质谱法检测计算得出人源瘦素的质量分数为58.2%,该结果与纯度扣除法相比,相对偏差为4.8%。由于纯度扣除法并非准确定量的方法,存在无法避免和校正的操作误差和系统误差,而同位素稀释质谱法是根据蛋白序列中水解得出的氨基酸来进行检测和计算,通过加入同位素标记物,利用括弧法,即高标-样品-低标-样品的顺序进样检测,可有效地降低样品前处理及仪器信号漂移对结果造成的影响,且在定量过程中通过准确称量和采用氨基酸国家标准物质,使检测结果可溯源至SI单位制,因此,同位素稀释质谱法具有更高的准确度。
2.6 人源瘦素含量测定结果的不确定度评价
2.6.1 人源瘦素相对分子质量的不确定度
人源瘦素由146个氨基酸构成,其中含有709个C原子,1 168个H原子,190个N原子、218个O原子和5个S原子。根据IUPAC给出的各元素的原子质量及其不确定度,可以计算出其标准不确定度,结果列于表3。
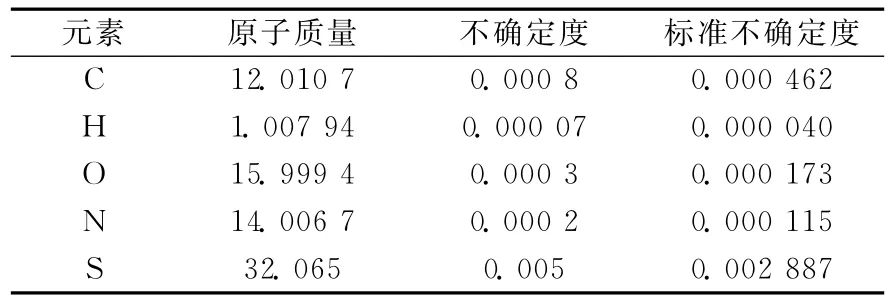
表3 人源瘦素中含有元素的原子质量及其标准不确定度Table 3 Standard uncertainties of the elements in human leptin
根据表3可计算人源瘦素蛋白摩尔质量的不确定度,示于式(5)。

该结果与人源瘦素的相对分子质量相比远小于0.1%,因此可忽略不计。
2.6.2 人源瘦素蛋白浓度c的相对标准不确定度ur(c)
1)由脯氨酸浓度计算得出的人源瘦素蛋白浓度值的相对标准不确定度ur(cpro)可由式(6)计算。
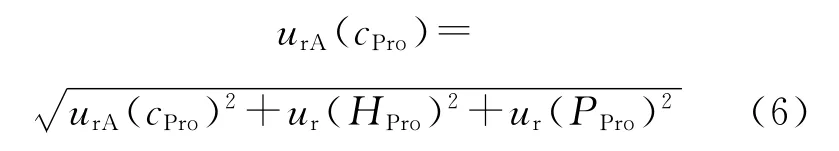
式中,urA(cPro)是脯氨酸浓度测量结果引入的A类不确定度,由为测量次(B类不确定度)是由水解效率引入的不确定度,由水解时间优化实验可知,当水解时间达到40h时,水解达到平衡,计算水解40、48、63、72 h所得的人源瘦素质量分数的RSD值为1%,即ur(HPro)为1%;ur(PPro)(B类不确定度)是由脯氨酸纯度标准物质引入的相对不确定度,由脯氨酸国家二级标准物质的标准证书得到其相对标准不确定度为1.5%。
经计算,ur(cPro)=2.0%。
需要说明的是,用精度为0.001mg的天平称量(0.5~1)mg样品,用精度为0.01mg的天平称量(100~200)mg样品,天平称量的相对不确定度小于0.1%,可忽略不计。
2)同上计算出缬氨酸和亮氨酸浓度值的相对不确定度ur(cVal)和ur(cLeu)。
3)人源瘦素蛋白浓度c的相对标准不确定度由式(7)计算:
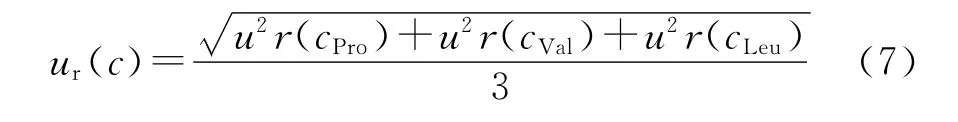
4)计算得出人源瘦素蛋白浓度c的相对标准不确定度为1.4%,结果列于表4。
2.6.3 人源瘦素的质量分数定值的不确定度
人源瘦素的质量分数A由式(3)计算得出,其中mLP和m水由精度为0.01mg的天平称量得出,称量质量为(25~200)mg,其不确定度小于0.1%,可忽略不计,得到其相对标准不确定度为ur(A)=ur(c)=1.4%。因此,人源瘦素蛋白质量分数的相对扩展不确定度为ur(A)=k×ur(A)=2×1.4%=2.8%(k=2),标准不确定度为u(A)=ur(A)×A=1.4%× 0.474=0.007(g/g),其扩展不确定度U(A)=u(A)×k=0.014g/g。可得到人源瘦素蛋白的质量分数为0.474g/g,其扩展不确定度为0.014g/g。所得扩展不确定度约为人源瘦素质量分数的2.9%,误差较小,表明利用高效液相色谱-同位素稀释-串联质谱法检测人源瘦素样品的结果较为准确。

表4 同位素稀释质谱法检测人源瘦素含量的相对标准不确定度Table 4 Standard uncertainties of human leptin quantified by IDMS
3 结论
建立了蛋白水解-高效液相色谱-同位素稀释-串联质谱法准确测定人源瘦素纯品绝对含量的方法。结合人源瘦素的氨基酸序列,选择了较为经济、稳定且易于购买的脯氨酸、亮氨酸和缬氨酸及其同位素标记物来定量人源瘦素样品。在110℃条件下,人源瘦素在6mol/L盐酸溶液中水解40h可达到完全水解。利用同位素稀释-液相色谱-串联质谱法检测瘦素的质量分数为0.582g/g,对定值结果进行不确定度评估,得到测量结果的扩展不确定度为0.014g/g(k=2),约为人源瘦素样品质量分数的2.9%,误差较小。与纯度扣除法检测人源瘦素的结果相比,相对偏差为4.8%。该定值方法可使人源瘦素蛋白的定值结果通过氨基酸溯源至SI单位,为人源瘦素标准物质的研究及标准测量方法的建立奠定了基础。
[1] CRÉPIN D,BENOMAR Y,RIFFAULT L,et al.The over-expression of miR-200ain the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment[J].Molecular and Cellular Endocrinology,2014,384(1/2):1-11.
[2] DALLONGEVILLE J,FRUCHART J C,AUWERX J.Pleiotropic hormone:Physiology,pharmacology,and strategies for discovery of leptin modulators[J].Journal of Medicinal Chemistry,1998,41(27):5 337-5 352.
[3] P W H,KASTIN A J.Leptin:A biomarker for sleep disorders[J].Sleep Medicin Reviews,2013,18(3):283-290.
[4] ABDALLA M,EFFAT D,SHETA M,et al.Serum leptin levels in rheumatoid arthritis and relationship with disease activity[J].The Egyptian Rheumatologist,2014,36(1):1-5.
[5] LEGGIO A,CATALANO S,DE MARCO R,et al.Therapeutic potential of leptin receptor modulators[J].European Journal of Medicinal Chemistry,2014,78:97-105.
[6] VONG L,YE C P,YANG Z F,et al.Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons[J].Neuron,2011,71(1):142-154.
[7] HAFFEJEAA F,NAICKER T,SINGH M,et al.Placental leptin in HIV-associated preeclampsia[J].European Journal of Obstetrics &Gynecology and Reproductive Biology,2013,171(2):271-276.
[8] GONZALEZ R R,LEAVIS P C.A peptide derived from the human leptin molecule is a potent inhibitor of the leptin receptor function in rabbit endometrial cells[J].Endocrine,2003,21(2):185-195.
[9] MANCOUR L V,DAGHESTANI H N,DUTTA S,et al.Ligand-induced architecture of the leptin receptor signaling complex[J].Molecular Cell,2012,48(4):655-661.
[10]FAN X F,CHEN Z H,HUANG Q,et al.Leptin as a marker for severity and prognosis of aneurismal subarachnoid hemorrhage[J].Peptides,2013,48:70-74.
[11]SONG C H,LIAO J,DENG Z H,et al.Is leptin a predictive factor in patients with lung cancer[J].Clin Biochem,2013,47(3):230-232.
[12]LIN M C,TSAI S Y,WANG F Y,et al.Leptin induces cell invasion and the upregulation of matrilysin in human colon cancer cells[J].Bio Medicine,2013,3(4):174-180.
[13]NEWMAN G,GONXSLEZ-PEREZ R R.Leptin-cytokine crosstalk in breast cancer[J].Molecular and Cellular Endocrinology,2014,382(1):570-582.
[14]LIU L L,WANG L J,ZHENG J D,et al.Leptin promotes human endometrial carcinoma cell proliferation by enhancing aromatase(P450arom)expression and estradiol formation[J].European Journal of Obstetrics &Gynecology and Reproductive Biology,2013,170(1):198-201.
[15]申玉星,全灿,马康.液相色谱-同位素稀释质谱法准确测定人血清中葡萄糖含量[J].质谱学报,2011,32(4):211-215.SHEN Yuxing,QUAN Can,MA Kang.The determination of glucose in human serum by liquid chromatography-isotopedilution mass spectrometry[J].Journal of Mass Apectrometry,2011,32(4):211-215(in Chinese).
[16]BARNIDGE D R,GOODMANSON M K,KLEE G G,et al.Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-MS/MS using protein cleavage and isotope dilution mass spectrometry[J].Journal of Proteome Research,2004,3(3):644-652.
[17]BONDAR O P,BARNIDGE D R,KLEE E W,et al.LC-MS/MS quantification of Zn-α2glycoprotein:A potential serum biomarker for prostate cancer[J].Clinical Chemistry,2007,53(4):673-678.
[18]WILLIAMS JR D K,MUDDIMAN D C.Absolute quantification of C-reactive protein in human plasma derived from patients with epithelial ovarian cancer utilizing protein cleavage isotope dilution mass spectrometry[J].Journal of Proteome Research,2009,8(2):1 085-1 090.
[19]KUHN E,WU J,KARL J,et al.Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and13C-labeled peptide standards[J].Proteomics,2004,4(4):1 175-1 186.
Quantification of Human Leptin by HPLC-IDMS
SUN Xue-qing1,2,HU Gao-fei1,SONG De-wei2,TIAN Ya-ping3,YAN Guang-tao3,XU Bei2,LI Hong-mei2
(1.Beijing University of Chemical Technology,Beijing100029,China;2.National Institute of Metrology,Beijing100013,China;3.Department of Clinical Biochemistry,PLA General Hospital,Beijing100853,China)
A method for the absolute quantification of human leptin was established by high performance liquid chromatography-isotope dilution mass spectrometry(HPLCIDMS).The hydrolysis conditions optimized were 40hwith 6mol/L HCl at 110℃.After hydrolysis,the sample was separated by HPLC,the transitions of proline(m/z116>m/z70)and proline-C5(m/z121>m/z 74),valine(m/z 118>m/z 72)and valine-C5
human leptin;absolutely quantification;isotope dilution mass spectrometry(IDMS);certified reference material(CRM);traceability
O 657.63
A
1004-2997(2015)01-0016-07
10.7538/zpxb.youxian.2014.0048
2014-02-08;
2014-05-08
科技部重大项目863计划(2011AA02A111);国家自然科学基金(21275134);科技部基础性工作专项(2011FY130100)资助
孙雪晴(1990—),女(汉族),河南人,硕士研究生,化学专业。E-mail:sunxq_89@163.com
宋德伟(1976—),男(汉族),黑龙江人,副研究员,从事蛋白质标准物质研究。E-mail:songdw@nim.ac.cn胡高飞(1977—),男(汉族),安徽人,副教授,从事色谱、波谱分析方法及应用研究。E-mail:hugf@mail.buct.edu.cn
时间:2014-08-20;
http:∥www.cnki.net/kcms/doi/10.7538/zpxb.youxian.2014.0048.html
(m/z123>m/z76),leucine(m/z 132>m/z 86)and leucine-D10(m/z 142>m/z 96)were monitored for quantitation by MRM mode.The content of human leptin was calculated to be 0.582g/g with the uncertainty of 0.014g/g(k=2).The calculated results are further validated by the purity deduction assay.It shows good consistency between the results of the two methods.In conclusion,application of this method for quantification of leptin will improve the precision,accuracy and lay the foundation for the future research of reference material of leptin.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
实用老年医学(2021年10期)2021-12-05
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
昆明医科大学学报(2021年3期)2021-07-22
商品与质量(2020年9期)2020-11-26
食品与生物技术学报(2020年2期)2020-01-05
祝您健康(2018年12期)2018-11-27
食品界(2018年8期)2018-09-03
浙江医学(2017年13期)2017-08-07
