
基于二噻吩并[2,3-b:3′,2′-d]噻吩开环反应的二芘基四联噻吩的制备及其激基缔合物荧光行为
2015-11-27 05:39李春丽王鹏飞
化学研究 2015年1期
徐 莉,仲 浩,李春丽,王鹏飞,王 华,*
(1.河南大学 化学化工学院,河南 开封 475004;2.河南大学 特种功能材料教育部重点实验室,河南 开封 475004)
二噻吩并[2,3-b:3′,2′-d]噻吩(DTT)是一类重要的有机光电功能材料,被应用于有机薄膜晶体管(Organic Thin Film Transistors,OTFTs)[1],同时作为有机合成的构筑模块应用于螺烯化学中的噻吩螺烯[2]与双螺烯[3]的合成.本课题组在研究该化合物的化学性质时首次发现该化合物可以在正丁基锂存在的条件下发生开环反应[4],同时发现除了正丁基锂外,其他强碱(如芳基锂)也可以与之发生开环反应[5].开环反应后产生芳硫基取代的联噻吩碳负离子(图1),如果遇到强的亲电试剂DMF,就可以产生结构特殊的芳硫基联噻吩甲醛化合物[5].但如果形成中间体芳硫基取代的联噻吩碳负离子时直接使用氯化铜氧化偶联,就可以设计合成出具有对称结构的二芳硫基取代的四联噻吩衍生物.我们关注芳基为芘基的衍生物,因为多年来芘基衍生物的光物理行为一直成为人们的关注对象,其原因在于芘基具有很好的发光能力,对环境的发光稳定性以及易形成激基缔合物(excimer)等特点[6].以芘基封端的线性低聚物[7]与支化聚合物[8]通过激基缔合物特征发光(发光峰位480nm)的变化,从而作为荧光探针表达出小分子、聚合物与生物分子[9]等分子形态.以噻吩低聚物作为桥链,连接两个芘基作为封端基团的化合物及其光物理行为的研究尚未见报道.本文作者基于DTT在芘基锂作用下的特殊的开环反应为出发点,通过对开环后的碳负离子中间体的直接氧化偶联获得了一种结构新颖的二芘基封端的四联噻吩(BPSQT)衍生物,并研究了该化合物的光物理行为,探讨了该化合物在溶液相与聚集态下的分子形态与发光行为,得到了一些有趣的结论.
1 实验部分
1.1 仪器与试剂
2-溴噻吩(濮阳惠成)、N-甲基丁二酰亚胺(国药集团)、无水氯化铜(Aldrich)、特丁基锂和正丁基锂(安耐吉)浓度参照文献标定[10],三氟乙酸(安耐吉)、无水乙醚(经金属钠钾处理后蒸馏)、无水四氢呋喃(经金属钠处理后蒸馏)、二(三甲基硅基)二噻吩并[2,3-b:3′,2′-d]噻吩(DTMS-DTT)按照文献方法制备[3a].
显微熔点测定仪(TX4-100,未经校准),Bruker 400MHz核磁共振谱仪,Trace DSQII(Finnigan)气相质谱联用仪(电子轰击能量70eV),AVATAR 360(Nicolet)傅立叶变换红外光谱仪,电喷雾离子源-高分辨质谱仪(Waters Micromass Q-Tof MicroTMSystem),Heλiosα(Unicam)紫外-可见分光光谱仪,SPEX-F212荧光光谱仪.
1.2 合成路线
BPSQT和模型分子PST的合成路线分别如图1和图2所示.

图1 BPSQT的合成路线Fig.1 The synthetic route to BPSQT

图2 模型化合物PST的合成Fig.2 The synthesis of model compound PST
1.3 2 ,2‴-二(1-芘基)-5,5′,5″,5‴-四(三甲基硅基)-3,3′:2′,2″:3″,3‴-四联噻吩(TTMSBPSQT)的合成
向25mL Schlenk瓶中加入1-溴芘(173.7mg,0.62mmol)与无水THF(7mL),于-78℃条件下加入n-BuLi(1.82mol/L in hexane,0.65mL,1.18 mmol)进行溴锂交换反应制备芘基锂溶液,并保持-78℃反应2h.另将DTMS-DTT(202.1mg,0.59 mmol)溶于7mL无水THF,降温至-78℃后用注射器将该溶液滴加到所制备的芘基锂溶液中,反应液由无色透明变成玫红色,继续在-50℃条件下反应3h.反应液降温至-78℃,加入CuCl2(204.6mg,1.49mmol),缓慢升至室温,搅拌过夜.0℃下用蒸馏水淬灭反应,用氯仿(2×20mL)萃取,蒸馏水(2×20mL)洗涤,有机相用无水硫酸镁干燥,经硅胶(300~400目)柱层析(淋洗液:V石油醚∶V氯仿=5∶1,石油醚沸程60~90℃)梯度淋洗得黄色固体产物TTMSBPSQT62.7mg,产率19.6%.熔点224~226℃.1H NMR(400MHz,CDCl3):8.43(d,J=9.2 Hz,2H),8.17(d,J=7.2Hz,2H),8.13(d,J=7.2Hz,2H),8.03~7.93(m,10H),7.73(d,J=8.0Hz,2H),7.10(s,2H),6.76(s,2H),0.23(s,18H),0.03(s,18H).13C NMR(100 MHz,CDCl3):143.80,141.37,140.61,137.70,136.93,136.23,135.82,133.39,133.09,131.36,130.97,129.87,129.14,127.77,127.39,127.27,127.13,126.04,125.20,125.13,125.10,124.96,124.47,123.87,-0.16,-0.47.MS(EI,70eV):m/z=1 082.2[M+];HRMS(EI):m/z calcd for C60H58Si4S6Na+(M+Na+):calc.:1 105.182 9,found:1 105.183 2.IR(KBr,cm-1):3 041,2 954.
1.4 2 ,2‴-二(1-芘基)-3,3′:2′,2″:3″,3‴-四联噻吩(BPSQT)的合成
将TTMSBPSQT(28.1mg,0.025 93mmol)溶于2mL氯仿中,加入三氟乙酸(TFA,0.1mL,1.36 mmol)进行反应,TLC薄层板跟踪反应至无原料,加水淬灭.反应液用3×15mL氯仿萃取,合并有机相,有机相分别用10mL水、10mL饱和碳酸氢钠溶液以及蒸馏水(2×10mL)洗涤,后经无水MgSO4干燥,硅胶(300~400目)柱层析(淋洗液:V石油醚∶V氯仿=3∶1,石油醚沸程60~90℃)分离得黄色固体BPSQT17.6mg,产率83.0%.熔点>300℃.1H NMR(400MHz,CDCl3):8.43(d,J=9.2Hz,2H),8.16(d,J=7.2Hz,4H),8.06(d,J=9.2 Hz,2H),8.03~7.94(m,8H),7.66(d,J=8.0 Hz,2H),7.36(d,J=5.2Hz,2H),7.21(d,J=5.2Hz,2H),7.06(d,J=5.2Hz,2H),6.83(d,J=5.6Hz,2H).13C NMR(100MHz,CDCl3):141.47,134.15,133.09,132.44,131.38,130.96,129.99,129.83,129.75,129.10,128.71,128.08,127.81,127.23,127.20,126.32,126.16,126.00,125.26,125.20,124.98,124.43,123.48.MS(EI,70eV):m/z=794.21[M+];HRMS(EI):m/zcalcd for C18H26S6(M+):calc.:794.035 9,found:794.036 3.IR(KBr,cm-1):3 040,2 924,2 854.
1.5 二(2-噻吩基)二硫醚(DTDS)的合成
向25mL Schlenk反 应 瓶 中 加 入2-溴 噻 吩(0.2mL,2.07mmol)与10mL无水乙醚,降温至-78℃,滴加n-BuLi(2.42mol/L in hexane,0.9 mL,2.18mmol),反应液为无色.反应2h后,加干燥的S粉(68.0mg,2.13mmol),升温至-30℃.反应2h后,于-30℃加入3mol/L盐酸(5mL)淬灭反应,反应液用2×20mL乙醚萃取,有机相分别用20mL水,20mL饱和碳酸氢钠溶液以及10 mL水洗涤.分离后的有机相用2×10mL饱和的铁氰化钾溶液与2×10mL水洗涤,有机相经无水硫酸镁干燥,硅胶(300~400目)柱层析(淋洗液:石油醚(沸程60~90℃))分离,得到黄色固体DTDS 174.4mg,产率73.1%.m.p.:56~57℃(文献值:56℃[11]).1H NMR(400MHz,CDCl3):7.52(dd,J=0.8Hz,J=7.2Hz,2H),7.18(dd,J=0.8 Hz,J=3.6Hz,2H),7.03(dd,J=3.6Hz,J=4.8Hz,2H).13C NMR(100MHz,CDCl3):135.64,132.28,127.74.MS(EI,70eV):m/z=229.82[M+].IR(KBr,cm-1):3 098,3 081.
1.6 1-芘基-2-噻吩基硫醚(PST)的合成
向25mL Schlenk反应瓶中加入1-溴芘(36.9 mg,0.13mmol)与7mL无水乙醚,降温至-78℃,加 入n-BuLi(2.42mol/L in hexane,0.06 mL,0.14mmol),保持在-78℃反应2h.滴加7 mLDTDS(30.0mg,0.13mmol)的无水乙醚溶液,在-78℃反应1h,自然升至室温,搅拌过夜.加水于0℃淬灭反应,反应液用3×15mL乙醚萃取,2×15mL水洗涤,有机相经硅胶(300~400目)柱层析(淋洗液:石油醚(沸程60~90℃))分离得到黄色固体PST 10.6mg,产率25.7%.m.p.:124~125℃.1H NMR(400MHz,CDCl3):8.69(d,J=9.2Hz,1H),8.23~8.17(m,3H),8.07~7.99(m,4H),7.89(d,J=8.0Hz,1H),7.44(dd,J=5.6Hz,J=1.2Hz,1H),7.37(dd,J=5.4Hz,J=1.2Hz,1H),7.08(qr,J=2.9Hz,1H).13C NMR(100MHz,CDCl3):134.78,132.84,132.15,131.37,130.95,130.47,130.40,129.48,128.14,127.86,127.71,127.53,127.22,126.23,125.43,125.36,125.14,125.08,124.48,123.68.MS(EI,70eV):m/z=316.02[M+].IR(KBr,cm-1):3 094,3 041,2 924,2 854.
2 结果与讨论
2.1 反应特点
基于本课题组对DTMS-DTT在不同有机锂试剂存在的条件下能够有效地发生开环反应[4-5],我们利用芘基锂对DTMS-DTT实施开环反应,形成芘硫基取代的联噻吩碳负离子PSBT-(图3),然后对中间体PSBT-碳负离子直接实施氯化铜氧化偶联,制备出具有对称结构的二芘硫基四联噻吩衍生物TTMSBPSQT.这是一种利用并三噻吩特殊的开环反应现象所拓展的制备四联噻吩低聚物的特殊方法,同时将两个具有发光功能的芘基作为封端基团连接在四联噻吩链上,形成了结构新颖的光电功能分子.此外,TTMSBPSQT分子结构上的四个TMS基团是可以高效去除的,通常使用三氟乙酸(TFA)[3a]或四 特 丁 基 氟 化 铵(TBAF)[12],本 文 采用TFA脱TMS,产率达83%.中间体TTMSBPSQT与产物BPSQT均通过了化合物结构的波谱鉴定.此外,我们设计合成了模型分子PST(见图2)作为光物理研究的对照材料.

图3 中间体TTMSBPSQT的形成机理Fig.3 The possible mechanism for formation of TTMSBPSQT
2.2 溶液中的吸收光谱
图4给出了化合物BPSQT、TTMSBPSQT与模型化合物PST在氯仿中的UV-Vis吸收光谱.

图4 化合物BPSQT,TTMSBPSQT与PST在氯仿中的UV-Vis光谱Fig.4 The UV-Vis spectra of BPSQT,TTMSBPSQT and PSTin chloroform
从图4中可以看出,BPSQT、TTMSBPSQT与模型化合物PST的吸收行为十分相近,分别在246、284和355nm处有3个吸收峰.将最短波长的吸收峰位(246nm)进行归一化处理,我们可以清楚地看出如下3个事实:其一,TTMSBPSQT分子中的TMS基团作为供电子基团并不能直接影响分子整体的吸收;其二,四联噻吩链对化合物整体吸收的贡献较小,与模型化合物PST相比较,四联噻吩链对BPSQT与TTMSBPSQT分子的最大吸收峰位没有贡献,仅在260~330nm的范围有一定的吸收;其三,与模型化合物PST的吸收行为相比较,BPSQT与TTMSBPSQT表现出最大吸收峰的展宽,并在370nm处出现肩峰.一方面说明了这3个化合物的吸收来源于芘硫基与相连接的噻吩之间的电子云共轭作用;另一方面可能由于四联噻吩链的折叠效应导致四联噻吩自身不能形成良好的共轭,特别是TTMSBPSQT分子中存在四个TMS基团,由于空间位阻因素而使得噻吩之间的共轭严重受阻.
2.3 溶液中的荧光光谱
图5给出了化合物BPSQT、TTMSBPSQT与模型化合物PST在室温下氯仿中的荧光光谱.当浓度为1.0×10-5mol/L,通常认为溶质分子间没有相互作用.图5所展示的荧光为单分子的荧光发射,可以明显地看出3种化合物之间存在荧光光谱上的差异,其中最为简单的是模型化合物PST,它的发射峰位在较短波方向上,其最大发射峰位在425 nm处,而在400nm与450nm处出现两个肩峰.由于芘基环与硫原子以及噻吩环之间存在共轭相互作用,其荧光光谱比芘分子自身有显著的红移与荧光光谱的展宽.
与模型化合物PST相比较,BPSQT与TTMSBPSQT的荧光发射峰呈现显著的红移现象,其在长波长方向上的最大发射峰均在470nm左右.其中BPSQT在短波长方向的发射峰在428nm左右,与模型化合物PST的最大发射峰位相一致.如果将BPSQT与PST的荧光发射光谱作一差谱,就可以得到图6,该荧光差谱的最大光谱峰位在482nm,这是典型的激基缔合物的荧光发射的位置[6].

图5 化合物BPSQT,TTMSBPSQT与PST在氯仿中的荧光光谱Fig.5 The fluorescene spectra of BPSQT,TTMSBPSQT and PSTin chloroform
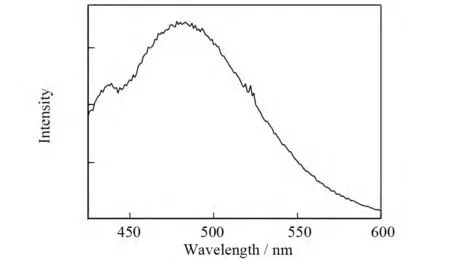
图6 化合物BPSQT和PST的荧光差谱Fig.6 The emission difference spectrum between BPSQTand PST
芘的激基缔合物的产生说明溶液中出现了两个芘基相遇的情况,并且是“三明治”的结构[6],两个芘基之间的相互作用的距离约为0.35nm.用于测试的样品TTMSBPSQT与BPSQT的浓度为1.0×10-5mol/L,我们也考察了其他低浓度的情况,如1.0×10-6mol/L,荧光的峰形并没有改变,说明了激基缔合物的产生来源于分子内的两个芘基之间的相互作用.我们进一步发现TTMSBPSQT比BPSQT更容易产生激基缔合物,因为TTMSBPSQT的荧光发射峰中激基缔合物的成分更大一些.这一现象说明了TTMSBPSQT与BPSQT分子在氯仿溶剂中存在两种构象的平衡,一种为舒展型(extended configuration),另一种为折叠型(folded configuration),其中折叠型构象在激发态下是可以发射分子内激基缔合物的荧光的(图7).

图7 溶液中BPSQT分子两种构象与THF-H2O二元溶剂体系中的聚集诱导发光现象Fig.7 Two configurations of BPSQTin solution and AIE-aggregation in THF-H2O binary solvent system
2.4 THF-H2O二元体系中的发光现象
图8给出了化合物BPSQT和PST在THF与水二元溶剂体系中的荧光发射光谱,样品的浓度保持1.0×10-5mol/L不变.在纯THF溶剂中,它们的荧光行为与其在氯仿中相一致.随着二元体系中水的体积分数从0逐步增加到60%时,介质的极性增大,但由于分子自身的极性比较小,特别是其中的BPSQT呈高度对称的结构,这使得分子受激后的偶极矩变化不大,因此荧光发射行为变化不大,说明这一过程没有造成分子构象的显著变化.当水的体积分数分别增加到大约70%(BPSQT)与90%(PST)时,化合物分子在二元溶剂体系中的溶解性明显降低,开始发生明显的分子之间的聚集,在480nm到500nm处产生新的荧光发射峰,并且荧光强度成倍增强.伴随水的体积分数的增加,最大发射峰的峰位也发生了一定程度的蓝移.这些现象说明由于分子间的聚集,在长波长方向上所产生的新的荧光发射峰是基于激发态的能量耗散途径变少,如溶剂化程度与溶质分子的布朗运动都显著下降,从而造成聚集态的荧光增强.这一现象与唐本忠教授提出的聚集诱导发光(AIE)的现象相一致[13]。所不同的是,他们提出的AIE现象是基于多个单键相连的噻咯[14-15]、四苯乙烯[16]等分子因分子间聚集时由于单键的自由旋转受阻降低了激发态的能量耗散的途径,从而造成了聚集态的荧光增强.而我们的体系是由于分子间的聚集,造成了分子间的激基缔合物的形成,产生了分子间激基缔合物的荧光发射.当分子间的聚集程度进一步增加,形成了多个芘基基团之间的相互作用,反而导致了聚集态发光强度的下降.其中,聚集态的发光呈现蓝移的现象可能与聚集体的极性下降有关.

图8 PST(上图)和BPSQT(下图)在THF和水的二元溶剂体系中的荧光光谱Fig.8 The fluorescene spectra of PST(top)and BPSQT(bottom)in THF-H2O binary solvent system
3 结论
综上所述,利用并三噻吩在碱性条件下的特殊开环反应,通过对开环产生的碳负离子的直接氧化偶联,从有机合成的新颖性角度制备了结构新颖的二芘硫基四联噻吩化合物.由于芘基基团之间存在较强的π-π相互作用,使得该功能分子表现出了舒展与折叠两种构象,在不良性的溶剂体系中能够展现出聚集诱导发光的现象,而这种发光是基于芘基之间形成的激基缔合物的特征发光.本研究工作对人们认识低聚噻吩为连接体的性质(如构象异构),以及拓展对芘基的激基缔合物的认知领域具有一定的理论价值.
[1]a)SHI J,XU L,LI Y,et al.Intermolecular interactions in organic semiconductors based on annelatedβ-oligothiophenes and their effect on the performance of organic field-effect transistors[J].Org Elect,2013,14(3):934-941.b)SHI J,LI Y,JIA M,et al.Organic semiconductors based on annelated boligothiophenes and its application for organic field-effect transistors[J].J Mater Chem,2011,8:17612-17614.c)ZHANG L,TAN L,WANG Z,et al.High-performance,stable organic field-effect transistors based on trans-1,2-(dithieno-[2,3-b:3′,2′-d]thiophene)ethene[J].Chem Mater,2009,21(9):1993-1999.
[2]a)RAJCA A,WANG H,PINK M,et al.Annelated heptathiophene:a fragment of a carbon-sulfur helix[J].Angew Chem Int Ed,2000,39(24):4481-4483.b)RAJCA A,MIYASAKA M,PINK M,et al.Helically annelated and cross-conjugated oligothiophenes:asymmetric synthesis,resolution,and characterization of a carbon-sulfur[7]helicene[J].J Am Chem Soc,2004,126(46):15211-15222.
[3]a)LI C,SHI J,XU L,et al.Syntheses and crystal structures of fused thiophenes:[7]helicene and double helicene,aD2-symmetric dimer of 3,3′-bis(dithieno[2,3-b:3′,2′-d]thiophene)[J].J Org Chem,2009,74(1):408-411.b)WANG Z,SHI J,WANG J,et al.Syn-theses and crystal structures of benzo hexathia[7]helicene and naphthalene cored double helicene[J].Org Lett,2010,12(3):456-459.c)LIU X,YU P,XU L,et al.Synthesis for the mesomer and racemate of thiophene-based double helicene under irradiation[J].J Org Chem,2013,78(12):6316-6321.
[4]WANG Z,ZHAO C,ZHAO D,et al.The preparation of substituted bithiophenyl aldehydes via the ring opening of dithieno[2,3-b:3′,2′-d]thiophene in the presence of n-BuLi[J].Tetrahedron,2010,66(12):2168-2174.
[5]ZHONG H,SHI J,KANG J,et al.Ring-opening reaction of 2,5-dioctyldithieno[2,3-b:3′,2′-d]thiophene in the presence of aryllithium reagents[J].Beilstein J Org Chem,2013,9:767-774.
[6]BAINS G,PATEL A,NARAYANASWAMI V.Pyrene:aprobe to study protein conformation and conformational changes[J].Molecules,2011,16(9):7909-7935.
[7]DE MELO S,COSTA T,FRANSCISCO A,et al.Dynamics of short as compared with long poly(acrylic acid)chains hydrophobically modified with pyrene,as followed by fluorescence techniques[J].Phys Chem Chem Phys,2007,9:1370-1385.
[8]DJHAMEL J.Internal dynamics of dendritic molecules probed by pyrene excimer formation[J].Polymers,2012,4(1):211-239.
[9]HUANG J,WU Y,CHEN Y,et al.Pyrene-excimer probes based on the hybridization chain reaction for the detection of nucleic acids in complex biological fluids
[J].Angew Chem Int Ed,2011,50(2):401-404.
[10]SUFFERT J.Simple direct titration of organolithium reagents using N-pivaloyl-o-toluidine and/or N-pivaloylo-benzylaniline[J].J Org Chem,1989,54(2):509-510.
[11]CHALLENGER F,MILLER S,GIBSON G.2-Thienyl-and 3-thienyl-thioacetic acids and their derivatives[J].J Chem Soc,1948:769-771.
[12]GOMMERMANN N,GEHRIG A,KNOCHEL P.Enantioselective synthesis of chiralα-aminoalkyl-1,2,3-triazoles using a three-component reaction[J].Synlett,2005(18):2796-2798.
[13]YU G,YIN S,LIU Y,et al.Structures,electronic states,photoluminescence,and carrier transport properties of 1,1′-disubstituted 2,3,4,5-tetraphenylsiloles[J].J Am Chem Soc,2005,127(17):6335-6346.
[14]LI Z,DONG Y,MI B,et al.Structural control of the photoluminescence of silole regioisomers and their utility as sensitive regiodiscriminating chemosensors and efficient electroluminescent materials[J].J Phys Chem B,2005,109(20):10061-10066.
[15]WANG W,LIN T,WANG M,et al.Aggregation emission properties of oligomers based on tetraphenylethylene[J].J Phys Chem B,2010,114(18):5983-5988.
[16]AN B,KWON S,JUNG S,et al.Enhanced emission and its switching in fluorescent organic nanoparticles[J].J Am Chem Soc,2002,124(48):14410-14415.
猜你喜欢
中国酿造(2022年1期)2022-02-07
同位素(2020年6期)2020-12-18
人民交通(2020年17期)2020-09-15
光谱学与光谱分析(2016年10期)2016-07-12
当代化工研究(2016年9期)2016-03-20
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
浙江化工(2014年9期)2014-08-15
应用化工(2014年9期)2014-08-10
无机化学学报(2014年9期)2014-02-28
