
融水小型猪线粒体DNA结构和系统进化的分析
2015-11-27 05:27:02徐嘉悦施赫赫王绪敏余细勇唐小江
中国比较医学杂志 2015年3期
徐嘉悦,施赫赫,王绪敏,高 翔,余细勇,于 军,唐小江
(1.中国科学院北京基因研究所,北京 100101;2.广东省医学实验动物中心,广东佛山 528248;3.广东贝格生物科技有限公司,广东佛山 528100;4.南京大学模式动物研究所,南京 210061;5.广东省医学科学院,广州 510060)
融水小型猪是源自广西苗寨的小黑猪,具有体型微小、性情温驯、产仔力强、母性较好等特点,具备培育成标准化实验用小型猪的有利特点。我们于2012年将该猪种源引到广东三水繁育,建立了基础种群和的系谱档案,已繁殖出F1代和F2代,参照北京市地方标准[1]初步建立了融水小型猪的质量控制标准。为了进一步掌握其遗传背景,本文对线粒体DNA进行了分析。
1 材料和方法
1.1 样本的收集和DNA的提取
融水小型猪由广东贝格生物科技有限公司(广东省医学实验动物中心小型猪基地)提供。用无菌肝素钠抗凝采血管取融水小型猪外周静脉血10 mL,用Qiagen公司的QlAamp DNA试剂盒按操作说明书提取DNA。
1.2 序列的测序和组装
采用全基因组鸟枪法的测序策略,使用Illumina基因组分析测序技术进行测序研究。为了提高组装的质量,对融水小型猪选取了插入片段大小为300 bp和500 bp的paired-end文库进行末端测序,用基于 Brujin图算法[2]的 SOAPdenovo软件(http://soap.genomeics.org.cn)对测序得到的短片段进行拼接组装[3-4],得到融水小型猪的完整线粒体DNA序列。
1.3 线粒体DNA的注释和分析
为了确定融水小型猪的线粒体结构,首先以Susscrofa的线粒体序列为参考序列,采用BLAST软件[4]完成了Susscrofa与融水小型猪的比对,确定线粒体的编码蛋白基因、rRNA和 tRNA,用软件CodonW对编码蛋白基因的密码子使用情况进行统计。另外用 tRNAscan-SE v.1.21软件[5]对线粒体tRNA的二级结构及各个tRNA的反义密码子进行预测。用clustal W[6]软件将融水小型猪的编码蛋白区序列与GenBank中的15头国内外的猪线粒体编码蛋白区序列(表1)进行比对。
用Dnasp5.10[7]软件计算实验材料的单倍型、遗传多样性和遗传分化的相关数值[8]。基于以上的单倍型聚类的分类,采用 Tajima’s D[9]和 Fu’s[10]两个中性检验模型对16个猪的单倍型分布进行中性检验,其中种群遗传格局及变异的检测在Arkequin3.11[11]中 实 施。 使 用 分 子 变 异 分 析(AMOVA)的方法以10000次重复随机抽样单倍型重排后进行显著性检验,以此来评价种群遗传变异水平与种群地理位置的相关性,使用统计量将遗传差异分为3个不同层次的地理等级。用Network 4.6(http://www.fluxus-engineering.com/sharenet.htm)对16个猪种进行聚类,并用MEGA 5.0[12]的最大似然法和邻接法对16个猪种的编码蛋白区进行系统进化分析。
2 结果
2.1 融水小型猪线粒体结构
融水小型猪与其他猪的结构框架相同,都是由13个编码蛋白,22个tRNA和2个rRNA组成(表2)。线粒体全长为16888 bp。线粒体DNA的核苷酸组成为A>C>T>G。线粒体的GC含量总是低于AT含量。
2.2 编码蛋白基因
融水小型猪由13个基因构成,除基因ND6位于线粒体的轻链上之外,其余12个基因包括ND1、ND2、ND3、ND4、ND4L、ND5、COX1、COX2、COX3、CYTB、ATP6、ATP8都在线粒体的重链(表2)。这13个基因的总长为11255 bp,其中基因ATP6和基因ATP8有42 bp的重叠区域,基因ND4L和基因ND4有7 bp的重叠区域,基因ND5和基因ND6有17 bp的重叠区域。位于融水小型猪重链的12个编码蛋白基因的碱基组分为A>C >T>G,其中ND1、ATP6、COX3、ND3、ND4、ND5、ND2,CYTB8 个基因的碱基组份是一致的:A >C >T >G,而基因 COX1、COX2、ATP8、ND4L的碱基组份为A>T>C>G;另外,位于融水小型猪轻链的ND6基因的碱基组份为C>A>T>G。
2.3 非编码蛋白区域:D-loop控制区和基因间区
在拼接的融水小型猪线粒体中,D-loop区的长度为1254 bp,tRNA-Pro和tRNA-Phe排布于D-loop区的两端。在D-loop区碱基组份为A>C>T>G,与线粒体DNA一致,AT的含量高于GC含量。此外,融水小型猪线粒体DNA中非编码的基因间区长度为654 bp,这些基因间区的主要原因是线粒体中tRNA簇的重排。其中最长的一段基因间区是位于ND1和tRNA之间的区域,长为139 bp。基因间区为高级别的系统进化研究提供了可能。
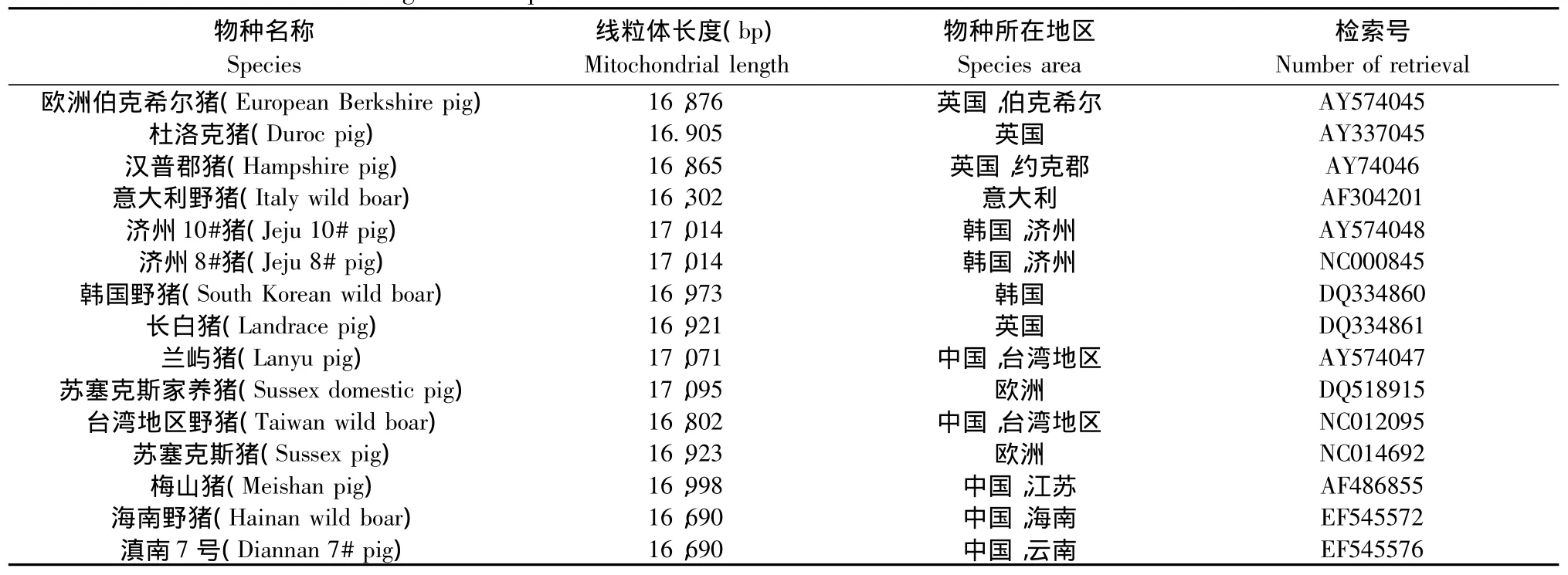
表1 15个猪种线粒体DNA的基本信息及其在NCBI数据库中的检索号Tab.1 List of mitochondrial genome sequences of Swine breeds and their accession numbers as retrieved from NCBI database
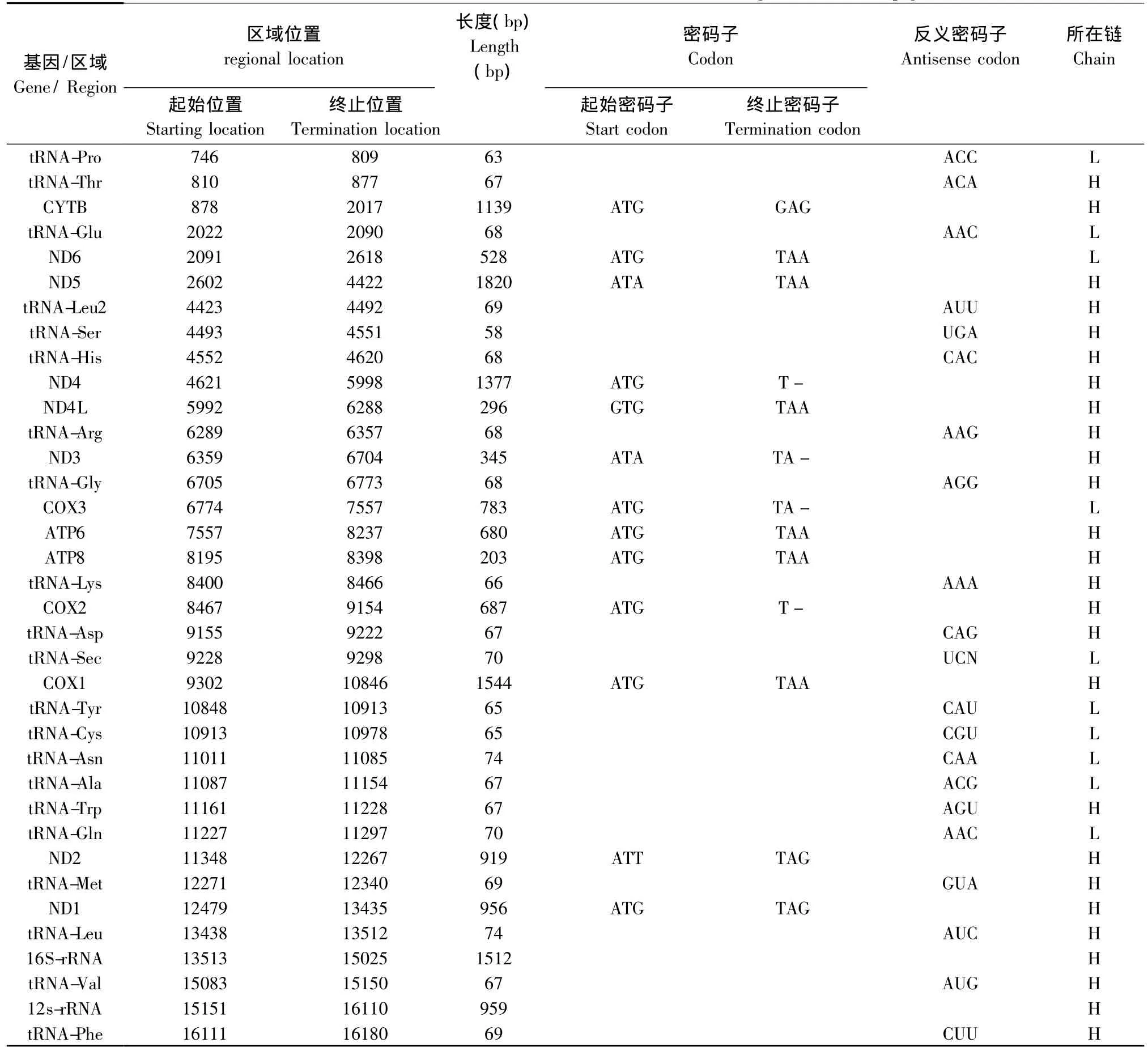
表2 融水小型猪线粒体DNA的结构特征Tab.2 Location of feature in the mitochondrial DNA of Rongshui miniature pig
2.4 核糖体RNA
线粒体中包括两个核糖体 RNA,分别为12S rRNA and 16S rRNA(表1),12S rRNA的长度为959 bp,16S rRNA的长度为1512 bp。16S rRNA编码于缬氨酸和亮氨酸之间,其碱基组份为A>T>C>G;12S rRNA编码于丙氨酸和缬氨酸之间,其碱基组份为A>C>T>G。12S rRNA和16S rRNA的GC含量均在40%左右,同样保持了GC含量低于AT含量这一特点。
2.5 转运RNA
对融水小型猪线粒体,我们预测了其22个转运RNA的二级结构,并确定了各转运RNA的反义密码子(如表2所示)。这22个转运RNA的GC含量都低于AT含量,其中精氨酸的GC含量最低,约21%;甲硫氨酸的GC含量最高,约47%。
融水小型猪线粒体DNA的密码子使用情况(表3)。包括终止密码子在内,共有3772个密码子,其中使用密码子CUA的数目最多,为306个,而AGG和CGG两个密码子没有被使用。在密码子的统计中,密码子第一位核苷酸使用的频率为A>C>T>G;而在密码子第二位置,碱基使用频率为T>C>A>G;在密码子的第三个位置,碱基使用频率为A>C>U>G。众所周知,密码子在不同位置使用频率的差异是由于密码子使用的偏好性造成的,上述的密码子使用情况也为研究线粒体DNA的进化提供了有用的信息[13]。
2.6 同义突变和非同义突变
同义突变和非同义突变之间的比例(Ka/Ks值)可用于分析不同物种之间的的系统进化关系[14]。通过对16个猪13个编码蛋白区的所有序列进行比对,构建了一个参考序列(Ref),分别计算每头猪的各个编码蛋白基因序列与Ref的Ka/Ks比值。结果如封2图1所示,16个猪种在编码蛋白区的Ka/KS值均小于1,说明所有13个编码蛋白基因都在承受着正选择,其中基因COX1的选择压力最大,这意味着该基因正在承受着很强的纯化选择;而ND5的选择压力相对较小,说明该基因是在进化中舍弃一些较为不利的突变。
2.7 系统进化分析
使用 MEGA软件中的 Kimura’s two-parameter模型(K2P)计算16个猪种的编码蛋白区的遗传距离。融水小型猪与其他猪的遗传距离最远,碱基的平均遗传距离为1.141。而对总体的遗传距离统计中,物种间以恒定速率进行核苷酸替换的标准差仅为0.0015。基于上述遗传距离矩阵,通过MEGA软件中的最大似然法和邻接法对16头猪的编码蛋白区构建系统进化树(图2),从图2可以看出,16个猪种可以分成四个类群,即欧洲群(E群)、亚洲1群(A1群)、亚洲2群(A2群)、亚洲3群(A3群)。其中,E群主要是由欧洲猪种组成,其中意大利的野猪是相对古老的猪种;A1群是由中国、日本、韩国等亚洲猪种及Berkshire猪共同组成,在这一分支中,也可以证明猪的进化是由亚洲起源后分化到欧洲的[15];A2群是由滇南7号、海南野猪和兰屿猪构成,这一结论也验证了之前的研究,即云南猪种和台湾地区兰屿猪是古老的猪种[15]。在系统进化分析中最值得关注的是融水小型猪,可以看出他比兰屿猪更为古老。
2.8 线粒体序列的单倍型分析及中性检验
系统进化分析是对过去已经发生的进化时间的模拟分析,为了验证系统进化分析结果的准确性,用单倍型的方法,对16个猪种的进化关系做了进一步分析。用Dnasp5.10[7]分析16个猪种的线粒体DNA的编码区序列,共得到14个单倍型,但单倍型的多样性指数为0.596。将分析后的单倍型数据利用Network软件作图(封2图3)。14个单倍型可以分成3大类:汉普郡猪、苏塞克斯家养猪、长白猪、苏塞克斯猪、杜洛克猪、意大利野猪、欧洲伯克希尔猪、聚为一类(类群1);济州8#猪、济州10#猪、韩国野猪、梅山猪和台湾地区野猪聚为一类(类群2);兰屿猪、滇南7号猪、海南野猪、融水小型猪聚为一类(类群3;其中类群1主要是欧洲猪种,类群2主要以亚洲的韩国和台湾地区猪种为主,类群3主要是中国4头古老的猪种。可以看出Network作图结果与系统进化分析的结果大致是一致的,这也进一步验证了融水小型猪是一个非常古老的猪种。
基于单倍型的分析结果,分别对3个类群进行了中性检验(如表4)。类群3不但具有较高的单倍型多样性(h=0.899),也具有较高的核苷酸多样性(Pi=0.03417),表明类群3中的猪种仍具有丰富的遗传多样性,存在着较高的进化潜力。对于类群1和类群2,单倍型多样性较高,但是核苷酸多样性很低。说明这两个类群的猪种经历过近期扩张或者进化历史很短[17]。本研究中类群1和类群3的Tajin a’s D与Fs值为负值且差异显著,这表明类群1和类群3中的猪种在历史上都发生了种群扩张。
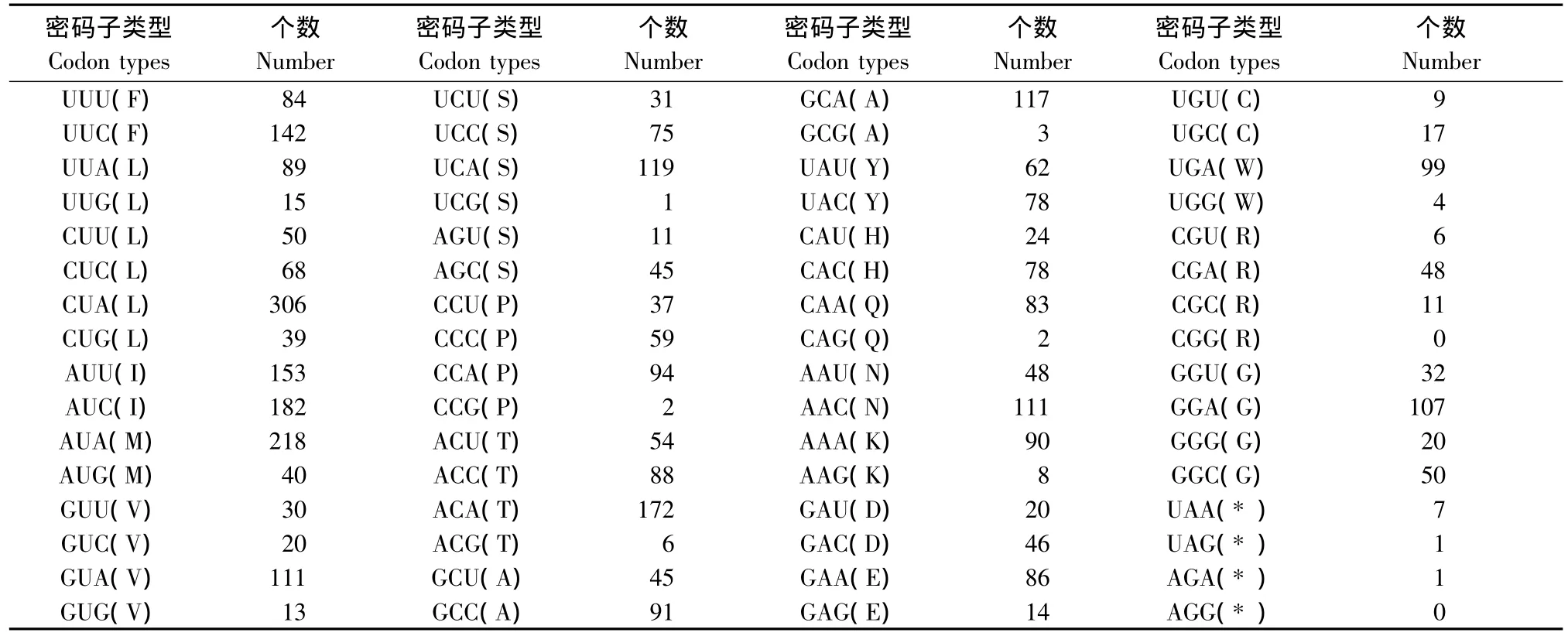
表3 融水小型猪密码子Tab.3 Codon usage for Rongshui miniature pig
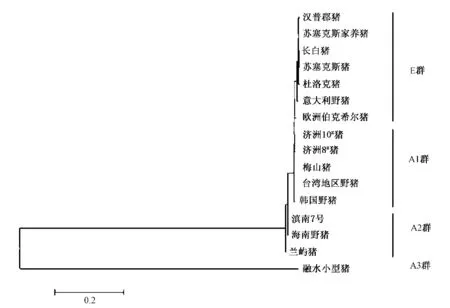
图2 16个猪种的系统进化分析(最大似然法和邻接法)Fig.2 The phylogenetic analysis of 16 pig breeds(NJ,ML analyses)
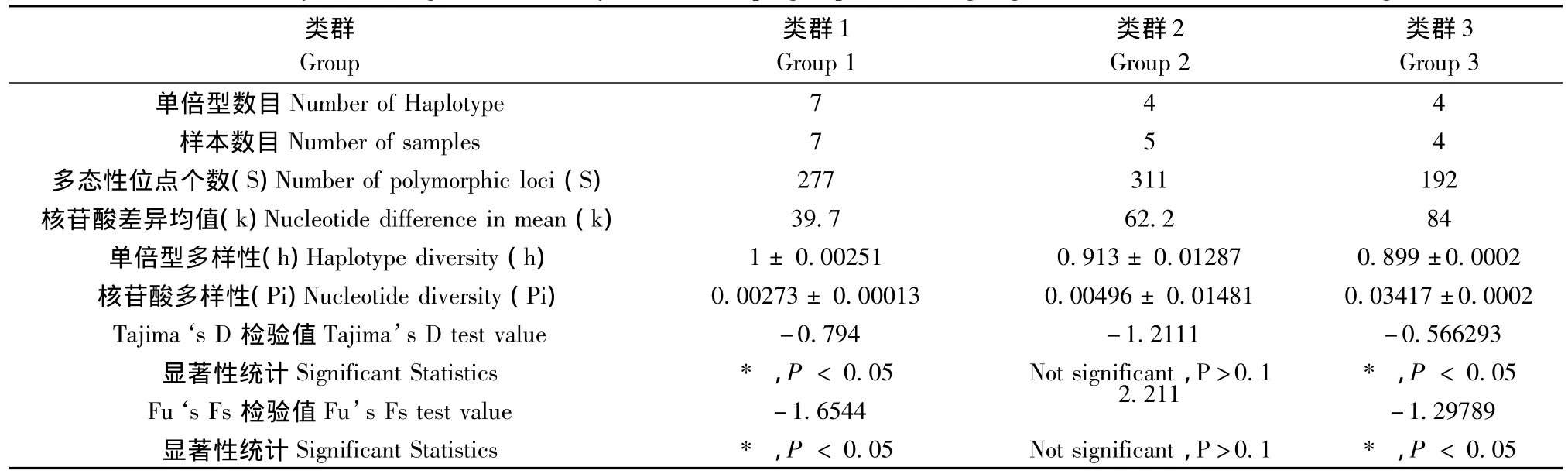
表4 对16个猪种编码蛋白基因的中性检验和遗传多样性分析Tab.4 Neutrality test and genetic diversity for main haplogroups in coding region for 16 swine mitochondrial genomes
3 讨论
线粒体DNA存在于细胞质线粒体机质中的闭合环状双链超螺旋DNA分子,是独立于细胞核染色体外的基因组,具有自我复制、转录和编码功能,是母系遗传,但同时受核 DNA的控制[2]。与核酸DNA相比,线粒体DNA具有分子结构简单,以母性方式遗传,核苷酸歧义大,进化速度快等特点。因此,使用线粒体研究哺乳动物群体遗传多样性和种内、种间亲缘关系等,一直受到很多学者的关注[15,18-20]。本研究采用新一代测序技术,对融水小型猪进行测序分析,拼接成完整的线粒体DNA序列,从分子遗传学角度评估了融水小型猪的结构质特性、系统进化关系及其遗传多样性。为融水小型猪的实验动物化提供科学依据。
随着新一代测序技术的发展,越来越多的学者对哺乳动物遗传机制的研究不再仅仅停留在核DNA水平上,而是更多的综合细胞质DNA进行分析。线粒体DNA结构简单、稳定,是母系遗传方式,在遗传过程中未曾发生重组。
本文使用新一代测序技术,用Illumina测序平台对融水小型猪进行从头测序,使用SOAPdenovo拼接成完整线粒体序列并进行注释。融水小型猪的线粒体DNA成环形,总长为16888 bp,由13个基因,22个tRNA和2个rRNA组成。其中GC的平均含量约为44%。线粒体的GC含量总是低于AT含量,这一结论与其他物种一致,如人的GC含量为44.4%[21],小鼠的 GC 含量为 26.6%[22]。本文结合公共数据库发布的15个猪种线粒体序列,以线粒体的编码蛋白区为研究对象,对猪种的遗传多样性进行了研究,进而对猪种的起源和进化进行探讨。从系统进化分析中我们可以得出,融水小型猪在系统进化树中成为一个新的独立外群,我们认为融水小型猪是一个古老于兰屿猪的传统中国猪种。为了进一步验证这个结论,我们对系统进化树中的三个类群进行了遗传多样性分析,Network的结果与系统进化分析的结果是吻合的。由滇南7号、海南野猪、兰屿猪和融水小型猪构成的类群3中有4个单倍型,其具有较高的单倍型多样性和核苷酸多样性显示出类群3具有丰富的遗传多样性。
Tajin a‘s D与Fu’s Fs中性检验常用以推测种群扩张的历史事件。如果Tajin a’s D与Fs值呈负值,且在统计学上达到显著标准,则说明序列中含有比中性进化模型中更多的核苷位点变化,可能预示着该种群曾经有过扩张的历史。融水小型猪的Tajin a’s D与Fs值为负值且差异显著,说明了类群3的古老猪种在长期进化过程中还在与其他的猪种还在发生基因交流和种群扩张。
另外,在系统进化分析时,兰屿猪、滇南7号猪、海南野猪与融水小型猪分成了不同的分支,而在Network分析中成为一个类群,出现这种现象的原因可能是融水小型猪在长期的进化过程中,发生了基因交流。中性检验的结果不但从统计水平上验证了融水小型猪在猪线粒体发展中的祖先地位,而且还解释了融水小型猪作为祖先猪种是如何与其他猪种发生基因流和种群扩张的。遗传多样性分析进一步提示融水小型猪是中国传统猪种中一种古老的小型猪。
本文对融水小型猪DNA进行了初步的分析,限于篇幅和GenBank的数据,未与哥廷根小型猪、西藏小型猪、巴马小型猪等其他实验用小型猪的线粒体DNA进行比较,有待进一步研究。
[1] 北京市质量技术监督局.实验用小型猪:微生物学等级及监测DB11T 828.1-2011[S];寄生虫学等级及监测 DB11T 828.2-2011[S];遗传质量控制 DB11T 828.3 -2011[S];病理学诊断规范 DB11T 828.4-2011[S];配合饲料 DB11T 828.5-2011[S];环境及设施 DB11T828.6 -2011[S],2011-11-10发布,2012-03-01实施.
[2] Pevzner,P.A.,H.Tang,M.S.Waterman,An Eulerian path approach to DNA fragment assembly[J].Proc Natl Acad Sci U S A,2001,98(17):74-85.
[3] Angiero and Natalie.,Pigs Prove to Be Smart,if Not Vain.The New York Times,2009.
[4] Altschul,S.F.,Gish,W.,Miller,W.,et al.,Basic local alignment search tool[J].J Mol Biol,1990,215(3):403-410.
[5] Lowe,T.M.S.R.Eddy,tRNAscan-SE:a program for improved detection of transfer RNA genes in genomic sequence[J].Nucleic Acids Res,1997,25(5):95 -98.
[6] Thompson,J.D.,D.G.Higgins,T.J.Gibson,CLUSTAL W:improving the sensitivity ofprogressive multiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice[J].Nucleic Acids Res,1994,22(22):73-80.
[7] Rozas,J.,Sanchez-DelBarrio,J.C.,Messeguer,X.,et al.,DnaSP,DNA polymorphism analyses by the coalescent and other methods[J].Bioinformatics,2003.19(18):49 -67.
[8] Kimura,M.,Molecular evolutionary clock and the neutral theory[J].J Mol Evol,1987.26(1-2):24-33.
[9] Tajima,F.,Statistical method for testing the neutral mutation hypothesis by DNA polymorphism[J].Genetics,1989,123(3):58-63.
[10] Fu,Y.X.,Statistical tests of neutrality of mutations against population growth,hitchhiking and background selection[J].Genetics,1997,147(2):91 -105.
[11] Excoffier L,L.L., Schneider S, Arlequin ver.3.0:an integrated software package for population genetics data analysis[J].Evolutionary Bioinformatics Online,2005,1:47 -50.
[12] Tamura,K.,Peterson,D.,Peterson,N.,et al.,MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolutionarydistance, and maximum parsimony methods[J].Mol Biol Evol.28(10):73 -91.
[13] Dubey,B., MeganathanP.R., HaqueI., DNA minibarcoding:an approach for forensic identification of some endangered Indian snake species[J].Forensic Sci Int Genet,5(3):18-24.
[14] Yang, Z. R. Nielsen, Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models[J].Mol Biol Evol,2000,17(1):32 -43.
[15] Chen,Huang.,H.L,Yang H.Y.,et al.Mitochondrial genome of Taiwan pig (Susscrofa) [J]. African Journalof Biotechnology,2011,10(13):2556-2561.
[16] Wu,G.S.,Yao,Y.G.,Qu,K.X.,et al.,Population phylogenomic analysis of mitochondrial DNA in wild boars and domestic pigs revealed multiple domestication events in East Asia[J].Genome Biol,2007,8(11):245 -253.
[17] Femandez,A.I.,Alves,E.,Ovilo,C.,et al.,Divergence time estimates of East Asian and European pigs based on multiple near complete mitochondrial DNA sequences[J].Anim Genet,42(1):86-88.
[18] Kijas,J.M.Andersson L.,A phylogenetic study of the origin of the domestic pig estimated from the near-complete mtDNA genome[J].J Mol Evol,2001,52(3):302 -8.
[19] Femandez,A.I.,Alves,E.,Ovilo,C.,et al.,Divergence time estimates of East Asian and European pigs based on multiple near complete mitochondrial DNA sequences[J].Anim Genet,42(1):86-89.
[20] Yang,S.,Zhang,H.,Mao,H.,et al.,The local origin of the Tibetan pig and additional insights into the origin of Asian pigs[J].PLoS One,6(12):218 -215.
[21] Anderson,S.,Bankier,A.T.,Barrell,B.G.,et al.,Sequence and organization of the human mitochondrial genome[J].Nature,1981,290(5806):45 -65.
[22] Bibb,M.J.,Van Etten,R.A.,Wright,C.T.,et al.,Sequence and gene organization of mouse mitochondrial DNA[J].Cell,1981,26(2 Pt 2):67 -80.
猜你喜欢
贵州畜牧兽医(2023年3期)2023-06-29 07:07:02
猪业科学(2022年10期)2022-11-03 09:46:04
音乐教育与创作(2022年3期)2022-04-26 03:42:44
猪业科学(2021年6期)2021-08-12 06:42:56
猪业科学(2021年3期)2021-05-21 02:06:24
新农民(2020年31期)2020-12-09 13:55:21
生物学通报(2020年11期)2020-10-22 01:20:20
中成药(2018年7期)2018-08-04 06:04:10
中学课程辅导·教学研究(2017年15期)2017-07-24 23:27:32
澳门月刊(2014年11期)2014-11-15 15:53:07
