
抗流感病毒药物研究进展
2015-11-27 14:59高岩刘宗英李卓荣
中国医药生物技术 2015年6期
高岩,刘宗英,李卓荣
·综述·
抗流感病毒药物研究进展
高岩,刘宗英,李卓荣
流感是由流感病毒引起的一种急性呼吸道传染病,具有流行面广、传染性强、发病率高等特点,其波及范围之广、造成经济损失之大位于传染性疾病之首[1]。近年来,随着全球物种活动范围的加大,人、禽、猪和其他种属间流感病毒基因重组的概率大大增加,使流感病毒变异性增强[2],产生了具有高致病性的新变种病毒[3]。目前,应对流感病毒感染的主要措施是疫苗接种和药物治疗。疫苗接种很难使老人、儿童等高危人群产生有效免疫,且不能为机体提供广泛持久的免疫防护;同时流感病毒株不断的进化和新病毒株的出现,更大大降低了疫苗的有效性[4];因此药物治疗仍然是最好的控制流感病毒传播的手段[5]。目前 FDA 已批准上市的抗流感病毒药物,包括 2 个神经氨酸酶抑制剂(奥司他韦和扎那米韦)和 2 个 M2 离子通道抑制剂(金刚烷胺和金刚乙胺),CFDA 除已批准上述 4 个药物上市外,还批准了一个神经氨酸酶抑制剂(帕拉米韦)上市。近十年来,这些抗流感病毒药物在体内外诱生出大量的耐药病毒株,如H1N1 病毒对奥司他韦产生了耐药、H1N1 病毒和 H3N2病毒对金刚烷胺产生了耐药[6],因此,设计研发新型抗流感病毒药物已迫在眉睫。
流感病毒是一种负螺旋单链 RNA 病毒,根据其核蛋白(nucleoprotein,NP)和基质蛋白(matrix protein,MP)抗原决定簇的不同,将流感病毒分为甲(A)、乙(B)、丙(C)3 个类型[7]。流感病毒的包膜上嵌有两种糖蛋白:血凝素(hemagglutinin,HA)和神经氨酸酶(neuraminidase,NA)。在流感病毒入侵宿主细胞及传播的过程中,这两种蛋白扮演着非常重要的角色[8]。流感病毒的复制周期主要包括以下几个阶段(图1):①黏附——病毒通过其表面的血凝素吸附在宿主细胞表面;②内吞——通过细胞内吞作用,病毒进入宿主细胞形成胞内体;③膜融合——病毒包膜与胞内体膜融合,病毒核糖核蛋白(vRNP)进入宿主细胞胞浆;④入核——病毒核糖核蛋白进入细胞核;⑤RNA 合成——在细胞核内进行病毒遗传信息的复制;⑥出核——合成且组装好的vRNP 被运送出细胞核;⑦组装——装配成成熟病毒,在宿主细胞表面出芽;⑧释放——神经氨酸酶水解唾液酸释放新病毒[9]。阻断流感病毒复制周期中的任何一个阶段,都可以有效地抑制病毒的感染。本文对近年来报道的作用于流感病毒复制周期中不同阶段的抗流感病毒化合物进行综述。

图1 流感病毒复制周期(1:黏附;2:内吞;3:膜融合;4:入核;5:RNA 合成;6:出核;7:组装;8:释放)
1 血凝素抑制剂
血凝素有两大主要功能:一是识别并结合宿主细胞表面的唾液酸受体,使病毒黏附于宿主细胞表面;二是在酸催化下发生构象转变,介导病毒包膜与胞内体膜融合,将病毒释放进入胞浆。血凝素抑制剂包括阻止病毒黏附于宿主细胞的黏附抑制剂和阻止病毒包膜与胞内体膜融合的膜融合抑制剂。
1.1 黏附抑制剂
1.1.1 唾液酸类似物 唾液酸(图2)是一类九碳单糖族化合物,在病毒与宿主细胞的结合和子代病毒从宿主细胞释放中均起重要作用,因此其类似物既可以是血凝素抑制剂,又可以是神经氨酸酶抑制剂[10]。游离的唾液酸单体在体内不能阻止病毒黏附,但多价态的唾液酸类似物可以有效地阻止病毒黏附于宿主细胞[11],如多价态的唾液酸偶联聚乙二胺(G4-SA)对于多种流感病毒亚型(H1N1、H2N2 和 H3N2)均具有抑制活性[12]。但多价态唾液酸类似物具有细胞毒性,Hendricks 等[13]合成了一系列连有脂质体的唾液酸类似物,这类化合物在弱化细胞毒作用的同时,具有较强的抗病毒活性。包埋摩尔百分比为 7.5% 唾液酸类似物的脂质体体外对H1N1 病毒血凝素的 IC90为 0.041 μmol/L;而且显著提高感染 1000 pfu H1N1 病毒(90% 致死量)小鼠的存活率。
1.1.2 抗病毒蛋白 最具代表性的抗病毒蛋白是唾液酸酶的融合蛋白,它能够移除宿主上皮细胞表面的唾液酸受体,从而抑制病毒的黏附。由于它不直接作用于病毒,因此不易出现病毒耐药性。DAS181 是一种广谱抗流感病毒药物,能够抑制包括高致病性禽流感 H5N1 在内的各种流感病毒。目前 DAS181 已经完成 II 期临床研究[14]。Cyanovirin-N(CV-N)是在蓝藻中发现的一种抗病毒蛋白,通过与病毒表面血凝素的甘露寡糖结合,降低病毒的感染性。CV-N 几乎对所有的甲型和乙型流感病毒株均表现出较高的抗病毒活性[15]。遗憾的是,细胞毒性和免疫原性阻断了该蛋白的进一步研发。近期从大肠杆菌细胞质中发现一种 linker-CVN(LCVN),即在 N 末端具有柔性亲水性多肽的 CV-N,后经过对其进行结构改造得到 N 端 α-胺聚乙二醇化的PEG20k-LCVN,其在体内外均表现出良好抗流感病毒活性,体外对 H3N2 和 H5N3 病毒的 EC50分别为 0.43 μmol/L和 0.04 μmol/L,明显优于阳性对照药利巴韦林;2.0 mg/(kg·d)给药剂量下可使感染 H3N2 病毒小鼠的存活率提高 2 倍[16]。
1.1.3 核苷类似物 2-脱氧尿苷(图2)在体外以剂量依赖型作用方式抑制 H1N1 病毒血凝素和神经氨酸酶,对H5N2 流感病毒的 IC50为 45 μg/ml,且在治疗剂量内无细胞毒作用。该化合物虽然靶向于病毒表面的血凝素和神经氨酸酶,但主要通过阻断糖基化过程发挥作用[17],作用机制新颖。
1.1.4 天然产物 Neoechinulin B(图2)是深海赤散囊菌F33 发酵产物的提取分离物,具有广谱抗流感病毒活性,对多种耐药病毒株(如耐奥司他韦 H1N1 病毒和耐金刚烷胺H3N2 病毒)有较强的抑制作用,EC50为 13~27 μmol/L。Neoechinulin B 能与血凝素结合,竞争性抑制血凝素与唾液酸的结合,阻止病毒黏附于宿主细胞[18]。该化合物不易产生耐药性,是具有研发前景的抗流感化合物。Ou 等[19]通过虚拟对接研究发现姜黄素(图2)也能与病毒血凝素结合,体外对 H1N1 流感病毒的 EC50为 0.17 μmol/L;其体内代谢产物主要为四氢姜黄素,能够通过与之不同的作用机制阻止病毒复制,说明姜黄素中的二烯酮结构是该类化合物表现出不同抑制作用的关键官能团。中药白芍醇提物中的脂溶性组分在体外对 H1N1 病毒的 IC50为 0.016 mg/ml[20],对H3N2 病毒也表现出良好的抗病毒活性,是一种对 H1N1、H3N2 以及耐奥司他韦流感病毒均有效的广谱抗病毒组分。有报道称,软羽衣草提取物具有抑制 H1N1、H3N2 和 H5N2流感病毒的活性[21]。另据报道,银杏叶提取物也能抑制血凝素与唾液酸结合,对 H1N1 和 H3N2 流感病毒有效[22]。
1.2 膜融合抑制剂
1.2.1 MBX2329 和 MBX2546 MBX2329 和 MBX2546(图3)是通过高通量筛选得到的具有广谱抗流感活性的化合物,能够抑制 2009 H1N1、禽流感 H5N1 以及耐奥司他韦 H1N1 病毒感染,与奥司他韦联合用药具有协同作用。作用机制研究表明,两个化合物均能抑制酸催化的血凝素构象转变,从而阻止血凝素介导的膜融合,对多种流感病毒(H5N1、H1N1 和耐奥司他韦 H1N1)的 IC50为 0.3~5.9 μmol/L。此类化合物可作为新型抗流感药物研发的先导化合物,也可用于研究酸催化血凝素构象转变的分子机制[23]。
1.2.2 苯磺酰胺类化合物 RO5464466 和 RO5487624(图3)是苯磺酰胺类化合物的代表,在体内外均能抑制 H1N1 病毒感染,在体外对不同 H1N1 流感病毒株的EC50为 0.09~0.66 μmol/L;在体内能够延长感染 40 倍半数致死量 H1N1 病毒(致死剂量)小鼠的存活时间,显著提高小鼠存活率;但该类化合物对 A/Hongkong/8/68 和A/Human/Hubei/3/2005 两株 H3N2 流感病毒无抑制活性。作用机制研究表明,这类化合物能够阻止低 pH 诱导的血凝素构象转变,是血凝素构象稳定剂。可能由于作用机制不同,此类化合物体内活性弱于阳性对照药奥司他韦[24]。
2 神经氨酸酶抑制剂
神经氨酸酶能够水解宿主细胞表面的唾液酸释放新病毒,是一个非常有吸引力的靶标。神经氨酸酶抑制剂具有对多种亚型流感病毒广谱有效、患者耐受性高等优点,得到广泛研究。
2.1 帕拉米韦
帕拉米韦(peramivir)(图4)是具有胍基和亲脂侧链的环戊烷类衍生物,是继扎那米韦和奥司他韦之后的第 3 种流感病毒神经氨酸酶抑制剂,其体外抗 H5N1 病毒的效果与扎那米韦和奥司他韦相当;感染 24 或 48 h 后注射单剂量药物,可大大提高感染 H1N1 病毒小鼠和感染 H5N1 病毒小鼠的存活率;具有耐受性好、毒性小等优点[25]。帕拉米韦注射剂能有效治疗耐奥司他韦的流感病毒感染,适用于流感危重患者和无法口服或吸入药物患者的治疗[26]。2013年4月,国家食品药品监督管理总局批准了抗流感新药帕拉米韦氯化钠注射液;该药在美国正在进行 III 期临床试验。
2.2 辛酸拉尼米韦
辛酸拉尼米韦(laninamivir octanoate)(图4)是神经氨酸酶抑制剂 R125489 的前药,通过单次吸入治疗流感。由于经鼻给药后其活性代谢产物拉尼米韦在呼吸道内滞留时间长,并且拉尼米韦可与各种类型流感病毒的神经氨酸酶发生稳定结合,因此具有长效抗流感病毒活性[27]。该药对甲型 H1N1、H3N2 流感病毒和乙型流感病毒的 EC50均达到纳摩尔级。目前该药已在日本上市,在美国正在进行 III期临床试验。相比于帕拉米韦而言,拉尼米韦更不易产生耐药性,而且有可能开发出静脉注射剂型,避免吸入给药的不顺应性。
2.3 奥司他韦类似物
AV5080(图4)对甲型流感病毒神经氨酸酶表现出强抑制活性,体外抗 H5N1 和 H1N1 流感病毒的 EC50分别为 0.03 和 0.07 nmol/L,而且对奥司他韦耐药流感病毒株也表现出强抑制活性,该化合物还显示出良好药代动力学特性[28]。AV5075S 是 AV5027(图4)的甲磺酸前药,是流感病毒神经氨酸酶的竞争性抑制剂[29]。AV5027 体外抗H1N1 流感病毒神经氨酸酶的 IC50为 0.18 nmol/L,是阳性对照药磷酸奥司他韦的 12 倍。对感染 H3N2 流感病毒小鼠的体内研究表明,以 5 mg/(kg·d) 的剂量给药,AV5075S的治疗效果优于阳性对照药奥司他韦,使小鼠存活率提高了80%~90%,消除了体重减轻效应,在该化合物的治疗剂量内没有观察到毒性反应[30]。Tamiphosphor(TP)(图4)也是奥司他韦衍生物,能与神经氨酸酶活性位点的 3 个精氨酸残基发生静电结合,具有较强的神经氨酸酶抑制活性,从而抑制人类和禽流感病毒的复制。另外,胍基-Tamiphosphor(TPG)(图4)对耐奥司他韦流感病毒株有效,EC50<1 μmol/L;鼻腔内给予低剂量的 TPG 能大大提高感染流感H1N1 病毒小鼠的存活率[31]。
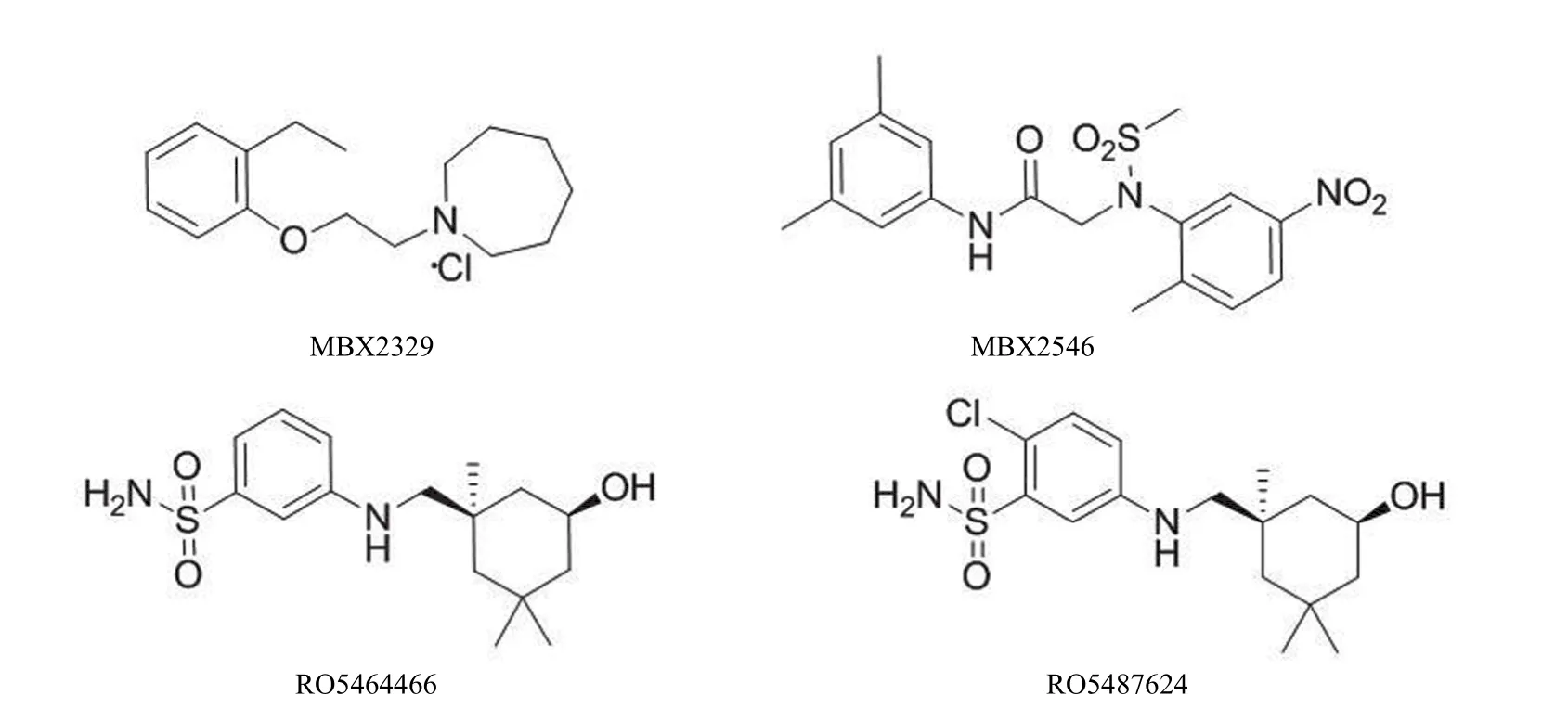
图3 膜融合抑制剂
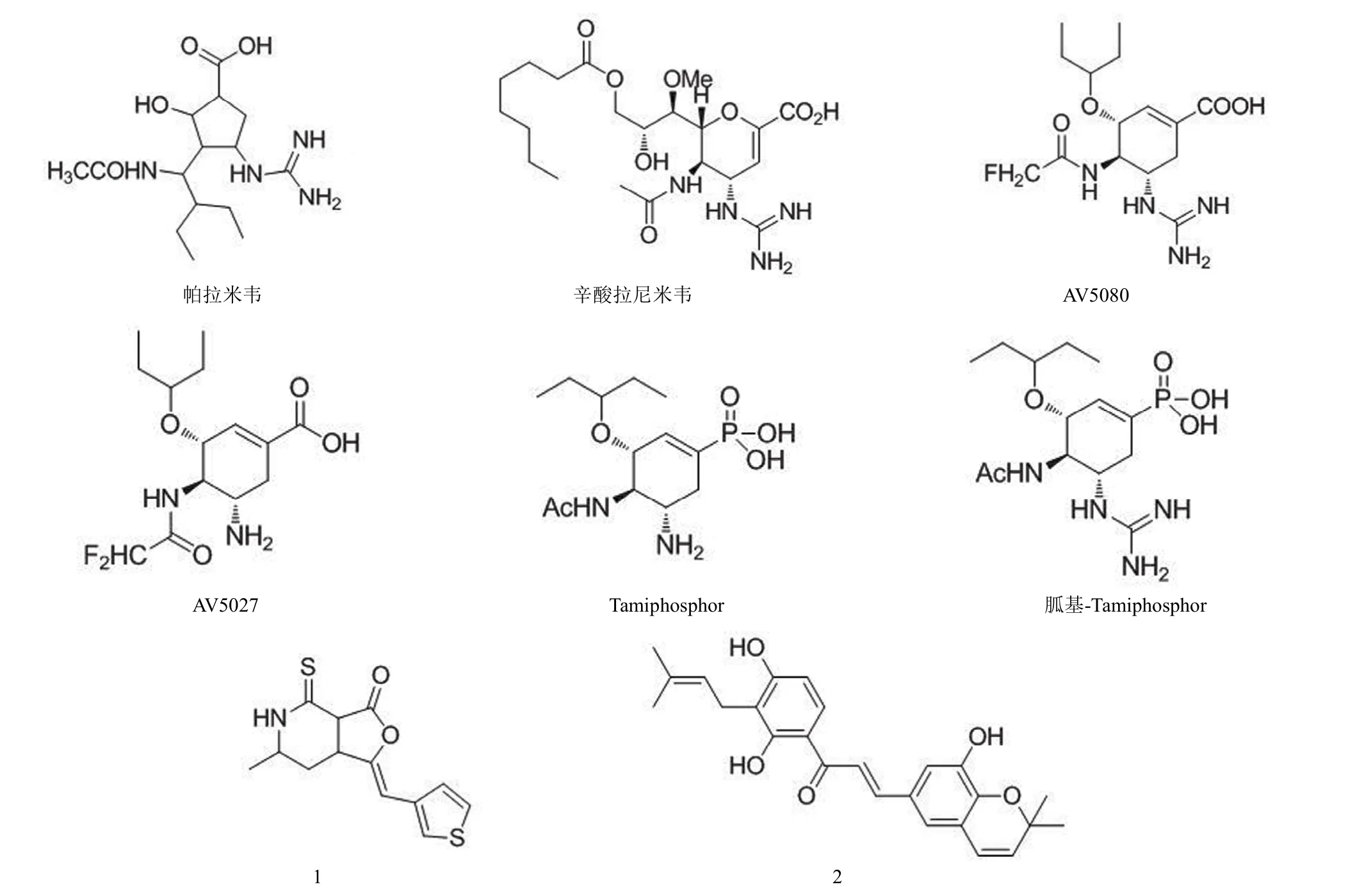
图4 神经氨酸酶抑制剂
2.4 二氢呋喃并吡啶酮类
Jang 等[32]通过对 7500 个化合物进行随机筛选和结构优化得到化合物 1(图4),其体外抗甲型 H1N1、H3N2 病毒和乙型流感病毒的 EC50为 5.0~6.2 μmol/L,并且浓度达到 900 μmol/L 时无细胞毒作用。化合物1以剂量依赖型作用方式降低感染细胞的病毒蛋白水平,也能以同种方式抑制子代病毒的产生。作用机制研究表明该化合物能抑制流感病毒的神经氨酸酶,阻止病毒释放。
2.5 天然神经氨酸酶抑制剂
Grienke 等[33]通过虚拟筛选发现了来自光果甘草的化合物 2(图4),其体外抑制 H1N1 病毒神经氨酸酶的 IC50为 0.25 μmol/L,该化合物可能通过抑制神经氨酸酶发挥抗流感病毒作用。也有报道将南非睡茄中的 Withaferin A(WA)与神经氨酸酶进行分子对接,两者具有较强的亲和力,且WA 具有弱化 H1N1 流感病毒神经氨酸酶的作用[34]。
3 核蛋白抑制剂
核蛋白的功能是与病毒 RNA 结合,通过相互连接包裹RNA 形成病毒核糖核蛋白,使病毒遗传信息得以复制。核蛋白具有高度保守性,是抗流感病毒药物理想的作用靶点[35]。
3.1 苦参喹诺里西啶类生物碱
从苦参属植物提取得到的苦参生物碱中,二氢苦豆碱(图5)抗甲型流感病毒活性最强,其体外抗 H1N1 流感病毒的 EC50为 11.2 μmol/L,对其进行化学修饰,得到了抗病毒活性更强的化合物 3(图5),其体外抗 H1N1 流感病毒的 EC50为 6.2 μmol/L。作用机制研究表明其能够作用于流感病毒复制周期的多个阶段,核蛋白是一个可能的作用靶标[36]。
3.2 萘普生
Lejal 等[37]通过虚拟筛选发现抗炎药萘普生(图5)能够与核蛋白结合,阻止核蛋白在病毒复制中发挥作用。其体外抗 H5N1 病毒的 EC50为 16 μmol/L,对 H1N1 和 H3N2突变株同样有效。另外,由于萘普生具有双重作用机制(抑制 COX-2 和核蛋白),疗效较高。
3.3 噻唑酰胺类化合物
Shen 等[38]研究发现阻断病毒核蛋白上的 E339…R416盐桥,能够阻止核蛋白形成聚合体,诱导单体核蛋白产生,致使其不能包裹 RNA 形成病毒核糖核蛋白。Shen 等针对E339…R416 盐桥建立了分子模型并进行虚拟筛选,发现化合物 4(图5)能够与该位点结合,其体外抗突变型 H1N1病毒的 IC50为 2.7 μmol/L。E339…R416 盐桥具有高度保守性,是研究抗病毒药物的一个较为理想的作用靶点。
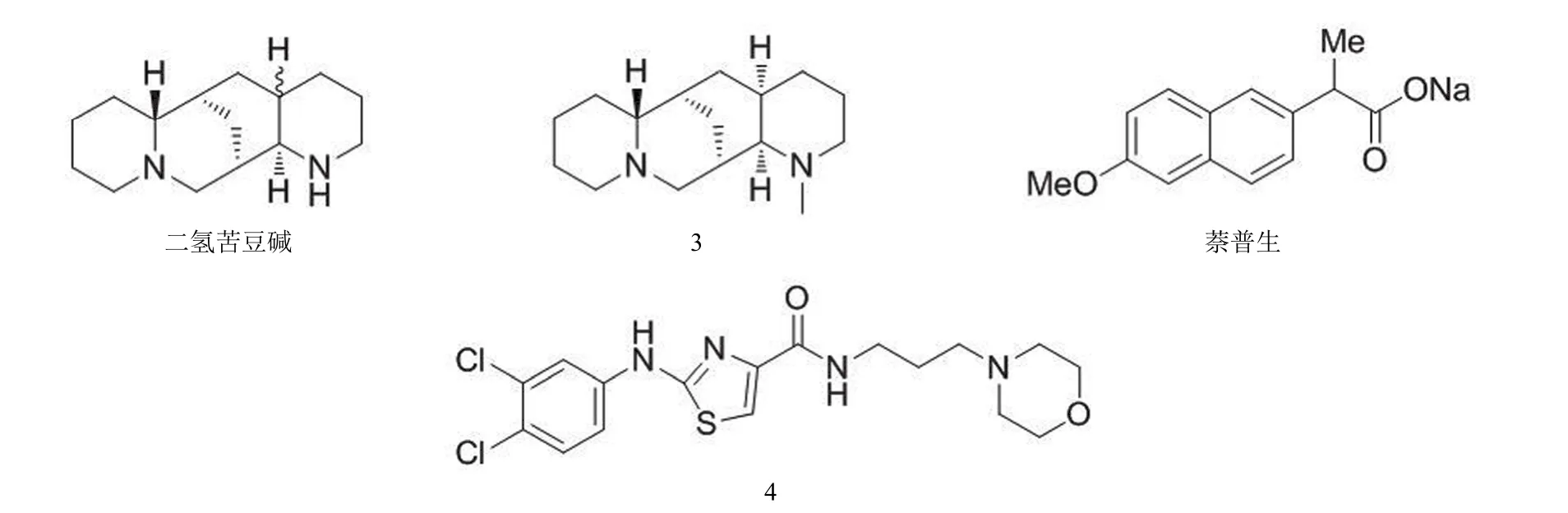
图5 核蛋白抑制剂
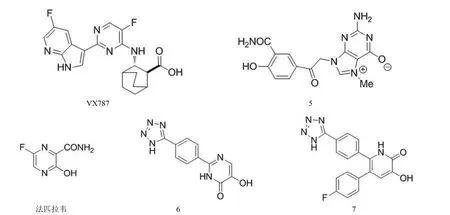
图6 RNA 聚合酶抑制剂
4 RNA 聚合酶抑制剂
流感病毒 RNA 依赖性 RNA 聚合酶(RdRP)负责宿主细胞中病毒 RNA 的转录和复制,由 PA、PB1 和 PB2三个亚基构成,其中 PA 具有核酸内切酶活性,参与病毒RNA 启动子的结合,且能与 PB2 相互作用;PB1 具有聚合酶和核酸内切酶活性;PB2 具有“抢帽”活性,与宿主细胞 mRNA 前体物结合,进行病毒 RNA 的转录。由于三个蛋白具有高度保守性,是非常有前景的抗流感药物作用靶点[39]。
4.1 法匹拉韦
法匹拉韦(favipiravir)(图6)为嘌呤核酸类似物,是一个特异性抑制病毒 RNA 聚合酶的抗流感病毒药物,通过被细胞磷酸核糖基化转变为活性形式法匹拉韦-RTP 发挥作用。法匹拉韦靶向于 RNA 依赖性 RNA 聚合酶,而宿主细胞无此靶标,因此该药不干扰宿主细胞 DNA 或 RNA的合成,无细胞毒作用。法匹拉韦为广谱抗流感药物,对包括 H1N1、H5N1、H7N9 流感病毒在内的多种甲型、乙型、丙型流感病毒均有抑制活性。法匹拉韦还能够抑制其他RNA 病毒,如沙粒病毒、布尼亚病毒以及非洲流行的埃博拉病毒等[40]。该药已在日本和美国完成抗流感病毒的 III期临床试验[41]。
4.2 环己基羧酸类化合物
Clark 等[42]发现环己基羧酸类化合物具有抗流感作用,其中 VX787(图6)对包括 2009 H1N1 和禽流感 H5N1 在内的各种甲型流感病毒均有较强抑制作用,其体外抗 H3N2病毒 EC50为 0.6 nmol/L;10 mg/kg·次(每日两次)给药可使感染 H1N1 病毒的小鼠肺部病毒量迅速减少[43]。VX787是 RNA 聚合酶 PB2 亚单位的抑制剂,其整体治疗效果优于阳性对照药奥司他韦,目前正在进行 II 期临床试验。
4.3 7-甲基鸟嘌呤衍生物
Pautus 等[44]通过在 PB2 的帽端结合腔进行分子对接,发现化合物 5(图6)能够与 PB2 帽端结合位点作用,抑制 PB2 的“抢帽”活性,从而使病毒不能利用宿主细胞mRNA 进行转录和复制。化合物 5 体外抗 H3N2 流感病毒 PB2 的 IC50为 0.6 μmol/L。
4.4 四氮唑类化合物
化合物 6[45]和 7[46](图6)是通过虚拟筛选发现的作用于 PA 的核酸内切酶抑制剂。两个化合物均具有较强抑制活性,其对 H1N1 流感病毒核酸内切酶的 IC50分别为0.15 和 0.011 μmol/L。
5 核糖核蛋白出核抑制剂
化合物 8(图7)是从牛角瓜乳胶中分离得到的一种新木脂素糖苷,其体外抑制人甲型和乙型流感病毒的 EC50为13.4~39.8 μmol/L,对禽流感病毒无抑制作用。初步构效关系研究表明,糖基部分连接的香草酚是活性必需基团。化合物 8 以剂量依赖型作用方式抑制流感病毒对核转位转录因子 NK-κB 通路的活化,阻断 vRNP 出核;同时核出口的病毒核糖核蛋白也被抑制[47]。由于化合物 8 作用机制独特,具有一定的研发前景。
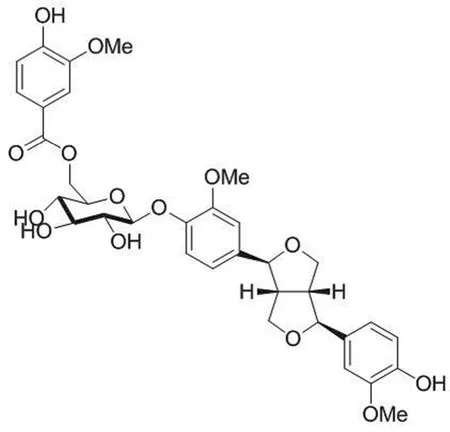
图7 核糖核蛋白出核抑制剂
6 结语与展望
新型流感病毒的大流行威胁着人类的健康,不断出现的病毒耐药性也敦促科学家们研发新型抗流感病毒药物。近几年报道了几种作用于流感病毒复制周期不同阶段的新型化合物,新型化合物抗病毒谱更广,对耐药病毒株的选择性更高。其中帕拉米韦和辛酸拉尼米韦则已经分别在我国和日本上市;DAS181、VX787 和法匹拉韦 3 个化合物已经进入临床研究阶段;有些化合物即将进入临床;多数化合物尚处于候选物研究阶段。相信随着人类对流感病毒复制周期分子机制认识的深入,随着新靶标的相继发现和确证,抗流感病毒药物的研究取得了较大进展,更多新型抗病毒药物将会被发现。
[1] Loregian A, Mercorelli B, Nannetti G, et al. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci, 2014, 71(19):3659-3683.
[2] Herfst S, Imai M, Kawaoka Y, et al. Avian influenza virus transmission to mammals. Curr Top Microbiol Immunol, 2014,385:137-155.
[3] Li C, Chen H. Enhancement of influenza virus transmission by gene reassortment. Curr Top Microbiol Immunol, 2014, 385:185-204.
[4] Lee YT, Kim KH, Ko EJ, et al. New vaccines against influenza virus. Clin Exp Vaccine Res, 2014, 3(1):12-28.
[5] Zhang Q, Zhao QS, Xiong RS, et al. Research progress of anti-influenza virus agents. Acta Phar Sinica, 2010, 45(3):289-299. (in Chinese)
张强, 赵庆生, 熊瑞生, 等. 抗流感病毒药物研究进展. 药学学报,2010, 45 (3):289-299.
[6] Hurt AC. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol, 2014, 8:22-29.
[7] Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol, 2014, 14(5):315-328.
[8] Oh DY, Hurt AC. A review of the antiviral susceptibility of human and avian influenza viruses over the last decade. Scientifica (Cairo), 2014,2014:430629.
[9] Sidorenko Y, Reichl U. Structured model of influenza virus replication in MDCK cells. Biotechnol Bioeng, 2004, 88(1):1-14.
[10] Suzuki K, Koyama T, Yingsakmongkon S, et al. Synthesis and biological evaluation of sialic acid derivatives containing a long hydrophobic chain at the anomeric position and their C-5 linked polymers as potent influenza virus inhibitors. Bioorg Med Chem,2012, 20(1):446-454.
[11] Wu WY, Jin B, Krippner GY, et al. Synthesis of a polymeric 4-N-linked sialoside which inhibits influenza virus hemagglutinin. Bioorg Med Chem Lett, 2000, 10(4):341-343.
[12] Landers JJ, Cao Z, Lee I, et al. Prevention of influenza pneumonitis by sialic Acid-conjugated dendritic polymers. J Infect Dis, 2002, 186(9):1222-1230.
[13] Hendricks GL, Weirich KL, Viswanathan K, et al. Sialylneolacto-N-tetraose c (LSTc)-bearing liposomal decoys capture influenza A virus. J Biol Chem, 2013, 288(12):8061-8073.
[14] Moss RB, Hansen C, Sanders RL, et al. A phase II study of DAS181, a novel host directed antiviral for the treatment of influenza infection. J Infect Dis, 2012, 206(12):1844-1851.
[15] Smee DF, Bailey KW, Wong MH, et al. Treatment of influenza A(H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res, 2008, 80(3):266-271.
[16] Wu C, Chen W, Chen J, et al. Preparation of monoPEGylated Cyanovirin-N's derivative and its anti-influenza A virus bioactivity in vitro and in vivo. J Biochem, 2015, 157(6):539-548.
[17] Krol E, Wandzik I, Gromadzka B, et al. Anti-influenza A virus activity of uridine derivatives of 2-deoxy sugars. Antiviral Res, 2013, 100(1):90-97.
[18] Chen X, Si L, Liu D, et al. Neoechinulin B and its analogues as potential entry inhibitors of influenza viruses, targeting viral hemagglutinin. Eur J Med Chem, 2015, 93:182-195.
[19] Ou JL, Mizushina Y, Wang SY, et al. Structure-activity relationship analysis of curcumin analogues on anti-influenza virus activity. FEBS J, 2013, 280(22):5829-5840.
[20] Ho JY, Chang HW, Lin CF, et al. Characterization of the anti-influenza activity of the Chinese herbal plant Paeonia lactiflora. Viruses, 2014,6(4):1861-1875.
[21] Makau JN, Watanabe K, Kobayashi N. Anti-influenza activity of Alchemilla mollis extract: possible virucidal activity against influenza virus particles. Drug Discov Ther, 2013, 7(5):189-195.
[22] Haruyama T, Nagata K. Anti-influenza virus activity of Ginkgo biloba leaf extracts. J Nat Med, 2013, 67(3):636-642.
[23] Basu A, Antanasijevic A, Wang M, et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion. J Virol, 2014, 88(3):1447-1460.
[24] Zhu L, Li Y, Li S, et al. Inhibition of influenza A virus (H1N1) fusion by benzene-sulfonamide derivatives targeting viral hemagglutinin. PLoS One, 2011, 6(12):e29120.
[25] Gu JF. Research progress on peramivir of novel anti-influenza virushighly potent neuraminidase inhibitor. Chin J New Drugs, 2013,22(9):989-997. (in Chinese)
顾觉奋. 新型抗流感病毒强效神经氨酸酶抑制剂帕拉米韦研究进展. 中国新药杂志, 2013, 22(9):989-997.
[26] Leang SK, Kwok S, Sullivan SG, et al. Peramivir and laninamivir susceptibility of circulating influenza A and B viruses. Influenza Other Respir Viruses, 2014, 8(2):135-139.
[27] Kakuta M, Kubo S, Tanaka M, et al. Efficacy of a single intravenous administration of laninamivir (an active metabolite of laninamivir octanoate) in an influenza virus infection mouse model. Antiviral Res,2013, 100(1):190-195.
[28] Ivachtchenko AV, Ivanenkov YA, Mitkin OD, et al. Novel oral anti-influenza drug candidate AV5080. J Antimicrob Chemother, 2014,69(7):1892-1902.
[29] Ivachtchenko AV, Ivanenkov YA, Mitkin OD, et al. A novel influenza virus neuraminidase inhibitor AV5027. Antiviral Res, 2013, 100(3):698-708.
[30] Ivachtchenko AV, Ivanenkov YA, Mitkin OD, et al. Novel oral anti-influenza prodrug candidate AV5075S. J Antimicrob Chemother,2014, 69(5):1311-1324.
[31] Chen CL, Lin TC, Wang SY, et al. Tamiphosphor monoesters as effective anti-influenza agents. Eur J Med Chem, 2014, 81:106-118.
[32] Jang YJ, Achary R, Lee HW, et al. Synthesis and anti-influenza virus activity of 4-oxo- or thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-ones. Antiviral Res, 2014, 107:66-75.
[33] Grienke U, Braun H, Seidel N, et al. Computer-guided approach to access the anti-influenza activity of licorice constituents. J Nat Prod,2014, 77(3):563-570.
[34] Cai Z, Zhang G, Tang B, et al. Promising anti-influenza properties of active constituent of withania somnifera ayurvedic herb in targeting neuraminidase of H1N1 influenza: computational study. Cell Biochem Biophys, 2015. [Epub ahead of print]
[35] Monod A, Swale C, Tarus B, et al. Learning from structure-based drug design and new antivirals targeting the ribonucleoprotein complex for the treatment of influenza. Expert Opin Drug Discov, 2015, 10(4):345-371.
[36] Dang Z, Jung K, Zhu L, et al. Identification and synthesis of quinolizidines with anti-influenza a virus activity. ACS Med Chem Lett, 2014, 5(8):942-946.
[37] Lejal N, Tarus B, Bouguyon E, et al. Structure-based discovery of the novel antiviral properties of naproxen against the nucleoprotein of influenza A virus. Antimicrob Agents Chemother, 2013, 57(5):2231-2242.
[38] Shen YF, Chen YH, Chu SY, et al. E339...R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc Natl Acad Sci U S A, 2011, 108(40):16515-16520.
[39] Tintori C, Laurenzana I, Fallacara AL, et al. High-throughput docking for the identification of new influenza A virus polymerase inhibitors targeting the PA-PB1 protein-protein interaction. Bioorg Med Chem Lett, 2014, 24(1):280-282.
[40] Oestereich L, Lüdtke A, Wurr S, et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res, 2014, 105:17-21.
[41] Furuta Y, Gowen BB, Takahashi K, et al. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res, 2013, 100(2):446-454.[42] Clark MP, Ledeboer MW, Davies I, et al. Discovery of a novel,first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J Med Chem, 2014, 57(15):6668-6678.
[43] Byrn RA, Jones SM, Bennett HB, et al. Preclinical activity of VX-787,a first-in-class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob Agents Chemother, 2015,59(3):1569-1582.
[44] Pautus S, Sehr P, Lewis J, et al. New 7-methylguanine derivatives targeting the influenza polymerase PB2 cap-binding domain. J Med Chem, 2013, 56(21):8915-8930.
[45] Sagong HY, Bauman JD, Patel D, et al. Phenyl substituted 4-hydroxypyridazin-3(2H)-ones and 5-hydroxypyrimidin-4(3H)-ones:inhibitors of influenza A endonuclease. J Med Chem, 2014, 57(19):8086-8098.
[46] Parhi AK, Xiang A, Bauman JD, et al. Phenyl substituted 3-hydroxypyridin-2(1H)-ones: inhibitors of influenza A endonuclease. Bioorg Med Chem, 2013, 21(21):6435-6446.
[47] Parhira S, Yang ZF, Zhu GY, et al. In vitro anti-influenza virus activities of a new lignan glycoside from the latex of Calotropis gigantea. PLoS One, 2014, 9(8):e104544.
10.3969/j.issn.1673-713X.2015.06.013
国家自然科学基金青年基金(81402827)
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所合成室
刘宗英,Email:liuzy@cde.org.cn
2015-07-16
猜你喜欢
广东农业科学(2022年11期)2023-01-13
基层中医药(2022年4期)2022-07-22
茶业通报(2021年4期)2022-01-21
科学与社会(2021年3期)2021-12-02
世界科学技术-中医药现代化(2021年5期)2021-11-05
大众健康(2021年4期)2021-05-06
世界科学技术-中医药现代化(2021年12期)2021-04-19
恋爱婚姻家庭·养生版(2020年3期)2020-04-13
科学24小时(2019年5期)2019-06-11
中成药(2017年7期)2017-11-22
